")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 10, Octobre 2008
|
|
|---|---|---|
| Page(s) | 841 - 846 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20082410841 | |
| Published online | 15 October 2008 | |
L’hyperactivité de la lipogenèse peut-elle conduire à la stéatose hépatique ?
Implication du facteur de transcription ChREBP
Can the hyperactivity of lipogenesis cause hepatic steatosis ? A role for ChREBP
Institut Cochin, Département d’Endocrinologie, Métabolisme et Cancer, Université Paris Descartes, CNRS (UMR 8104), Paris, France
Inserm, U567, 24, rue du Faubourg Saint Jacques, 75014 Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
L’obésité, la résistance à l’insuline et le diabète de type 2 s’accompagnent fréquemment d’une accumulation excessive de lipides (stéatose) dans des tissus qui, normalement, n’en stockent pas à long terme. C’est en particulier le cas du foie qui joue un rôle central dans le maintien de l’homéostasie énergétique. Ainsi, la prévalence de la stéatose hépatique est en augmentation constante dans nos sociétés industrialisées. La découverte du facteur de transcription ChREBP a permis des avancées considérables dans la compréhension du contrôle du métabolisme glucido- lipidique. Ce facteur de transcription, qui relaie l’action du glucose sur l’expression des gènes, apparaît comme un déterminant central de la physiopathologie de la stéatose hépatique et de la résistance à l’insuline chez la souris. Bien que son importance chez l’homme reste à démontrer, ChREBP pourrait constituer une cible thérapeutique importante.
Abstract
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease associated with insulin resistance, obesity and type 2 diabetes. Excessive accumulation of triglycerides (TG) is a hallmark of NAFLD and therefore, a better understanding of the steps involved in regulating hepatic TG synthesis might yield novel information regarding the prevention and treatment of NAFLD. In the recent years, the transcription factor ChREBP has emerged as a major mediator of glucose action on lipogenic genes and as a key determinant of lipid synthesis in vitro. More importantly, this factor has been described to play a central role in hepatic steatosis and insulin resistance physiopathology. Although its implication in human disease has not yet been demonstrated, ChREBP could be an interesting therapeutic target against metabolic syndrome components.
© 2008 médecine/sciences - Inserm / SRMS
La prévalence de la stéatose hépatique non alcoolique est en augmentation constante dans les pays industrialisés. Cette pathologie, qui se caractérise par une accumulation excessive de lipides dans le foie, est fortement associée au syndrome métabolique, défini par l’existence de troubles tels qu’une obésité abdominale, une résistance à l’insuline avec ou sans hyperglycémie, une dyslipidémie et une hypertension [1]. La stéatose hépatique fait partie d’un large spectre d’atteintes hépatiques regroupées sous le terme de NAFLD (non alcoholic fatty liver disease) ou stéato-hépatopathies non alcooliques. Parmi les NAFLD, on distingue ainsi la simple stéatose et les NASH (non alcoholic steato-hepatitis), caractérisées par une stéatose associée à la présence de lésions inflammatoires évolutives responsables de l’apparition d’une fibrose et d’une véritable cirrhose pouvant se compliquer de carcinome hépatocellulaire. Il a été proposé en 1998 un modèle permettant d’expliquer la pathogénie des NAFLD. Selon ce « modèle des 2 attaques » [2], l’accumulation de lipides dans le foie constitue une première phase adaptative qui fragilise les cellules, les rendant ainsi plus sensibles aux agressions telles que le stress oxydatif, la peroxydation lipidique et certaines cytokines conduisant à l’inflammation et à la fibrose. Ainsi, bien que chez certains individus, la stéatose demeure bénigne, elle évolue en stéato-hépatite chez 10 à 20 % des patients obèses et résistants à l’insuline [3].
Lors du développement de la stéatose hépatique, les lipides qui s’accumulent dans le foie sont majoritairement des triglycérides, issus de l’estérification de trois acides gras avec un glycérol-3-phosphate. Les acides gras utilisés pour la synthèse des triglycérides proviennent non seulement du pool plasmatique d’acides gras non estérifiés (issus de la lipolyse du tissu adipeux), mais également des acides gras néosynthétisés à partir du glucose par la voie de la lipogenèse hépatique (Figure 1). Les triglycérides hépatiques sont ensuite stockés dans des gouttelettes lipidiques ou sécrétés dans le sang sous forme de lipoprotéines de très faible densité (VLDL, very low density lipoproteins). Les triglycérides peuvent également être hydrolysés dans l’hépatocyte et les acides gras qui en résultent servent de substrats pour la β-oxydation ou sont réestérifiés et engagés dans la voie de sécrétion des VLDL (Figure 1).
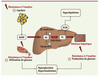 |
Figure 1. Étapes métaboliques conduisant au développement de la stéatose hépatique. Lors du développement de la stéatose hépatique, les lipides qui s’accumulent dans le foie sont majoritairement des triglycérides (TG). En raison de l’état de résistance à l’insuline caractéristique du patient obèse pré-diabétique, la lipolyse au niveau du tissu adipeux n’est plus efficacement inhibée par l’insuline et des concentrations importantes d’acides gras non estérifiés (AGNE) sont libérées dans la circulation sanguine. Ces acides gras (AG) sont captés par le foie. En parallèle, et de manière paradoxale, la synthèse de novo d’acides gras par la voie de la lipogenèse est stimulée par l’hyperglycémie et l’hyperinsulinémie. La combinaison de l’augmentation de la voie de la lipogenèse et de la diminution de la voie de β-oxydation des acides gras conduit au développement de la stéatose hépatique. Les TG hépatiques sont stockés dans des gouttelettes lipidiques ou sécrétés dans le sang sous forme de lipoprotéines de très faible densité (VLDL). L’accumulation excessive des concentrations de TG hépatiques exacerbe le phénotype de résistance à l’insuline dans le foie, conduisant à l’augmentation de la production hépatique de glucose. Au niveau du muscle squelettique, les concentrations élevées d’AGNE conduisent à une résistance à l’insuline et à une diminution de l’utilisation de glucose dans ce tissu. |
Une altération de chacune de ces étapes métaboliques (excès de captage par le foie, augmentation de la synthèse de novo d’acides gras et/ou défaut d’export et de β-oxydation) peut contribuer au développement de la stéatose hépatique (Figure 1). Des données récentes obtenues chez l’Homme suggèrent que la lipogenèse jouerait un rôle important dans le développement de la stéatose hépatique [4, 5] et contribuerait pour environ 30 % au stockage de triglycérides dans le foie des patients atteints de la maladie [6].
La synthèse de novo d’acides gras dans le foie par la voie de la lipogenèse
La lipogenèse est la voie métabolique qui permet de synthétiser des acides gras à partir du glucose. Elle nécessite le métabolisme du glucose en pyruvate par la voie de la glycolyse permettant ainsi de fournir les carbones nécessaires à la synthèse des acides gras (Figure 2). L’activité des voies de la glycolyse et de la lipogenèse est étroitement contrôlée par les conditions nutritionnelles. Ainsi, un repas riche en hydrates de carbone stimule la synthèse de lipides alors que le jeûne ou un repas riche en lipides l’inhibe. Les enzymes impliquées dans ces voies incluent la glucokinase (GK) et la pyruvate kinase (L-PK) pour la glycolyse, l’acétyl-CoA carboxylase (ACC) et la fatty acid synthase (FAS) pour la lipogenèse (Figure 2). La majorité de ces enzymes est contrôlée à court terme par des mécanismes post-traductionnels et allostériques, mais la principale régulation s’effectue à long terme au niveau transcriptionnel (pour revue, voir [7]).
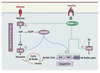 |
Figure 2. Contrôle transcriptionnel des voies de la glycolyse et de la lipogenèse. En réponse à une ingestion d’hydrates de carbone, l’augmentation de la glycémie stimule la sécrétion d’insuline par les cellules β du pancréas endocrine. Dans le foie, l’insuline active l’expression des gènes de la glycolyse et de la lipogenèse, nécessaires à la synthèse de lipides. L’effet transcriptionnel de l’insuline est transmis par le facteur de transcription SREBP-1c qui stimule la glucokinase (GK), les enzymes de la lipogenèse [l’acétyl-CoA carboxylase (ACC), la fatty acid synthase (FAS)], ainsi que la stéaroyl-CoA désaturase 1 (SCD-1) qui catalyse la synthèse d’acides gras mono-insaturés et la glycéraldéhyde 3-phosphate acyltransférase (GPAT) qui permet la synthèse de triglycérides (TG). L’action du glucose est quant à elle relayée par le facteur de transcription ChREBP, qui, induit par le xylulose 5-phosphate (Xu5P), un intermédiaire métabolique de la voie des pentoses, stimule l’expression de la L-pyruvate kinase (L-PK) et agit en synergie avec SREBP-1c pour induire les gènes de la lipogenèse. G6P : glucose 6-phosphate ; PEP : phosphoénolpyruvate. |
Contrôle de la lipogenèse par l’insuline : implication du facteur de transcription SREBP-1c
En réponse à une ingestion d’hydrates de carbone, l’augmentation de la glycémie stimule la sécrétion d’insuline par les cellules β du pancréas endocrine. Au niveau du foie, l’insuline active l’expression des gènes nécessaires à la synthèse de lipides. Une équipe française a clairement démontré que les effets géniques de l’insuline sur les gènes de la glycolyse (en particulier la GK) et de la lipogenèse sont relayés par le facteur de transcription SREBP-1c (sterol regulatory element binding protein) [8] (Figure 2). Le rôle clé de SREBP-1c dans la lipogenèse a été établi chez des souris surexprimant la forme active de ce facteur dans le foie. Ces souris développent une stéatose massive due à l’activation de l’expression des gènes lipogéniques [9, 10]. Cependant, dans le foie des souris invalidées pour SREBP-1c, bien que l’expression des gènes de la lipogenèse soit diminuée de 50 % [11], une activité lipogénique indépendante de SREBP-1c persiste en réponse à la réalimentation, suggérant l’existence d’un second niveau de contrôle. De plus, la L-PK, une enzyme clé de la glycolyse, est contrôlée exclusivement par le glucose [12], indépendamment de SREBP-1c [13], indiquant que le glucose per se agit comme une molécule signal (Figure 2).
Identification du facteur de transcription ChREBP comme médiateur de l’action du glucose
Jusqu’à récemment, la voie de signalisation initiée par le glucose pour activer la transcription des gènes cibles n’était pas connue. L’identification d’éléments de réponse aux hydrates de carbone (ChoRE) dans le promoteur des gènes sensibles au glucose suggérait l’existence d’un facteur de transcription relayant ces effets. La découverte du facteur de transcription ChREBP (carbohydrate responsive element binding protein) par le groupe de Uyeda en 2001 a permis de confirmer cette hypothèse. ChREBP a été caractérisé à partir d’extraits nucléaires de foies de rats nourris avec un régime riche en hydrates de carbone pour sa capacité à interagir avec le ChoRE de la L-PK [14]. ChREBP est une protéine de 94,6 kDa dont la structure est très conservée entre les espèces, avec une homologie de séquence de 82 % entre l’homme, le rat et la souris [15]. Ce facteur de transcription contient plusieurs domaines fonctionnels dont un signal de localisation nucléaire (NLS), un domaine de liaison à l’ADN de type bHLH-LZ (basic domain, helix-loop-helix, leucine zipper) et des domaines riches en proline qui interviennent dans les interactions protéine-protéine.
Afin de confirmer le rôle de ChREBP en tant que médiateur majeur de l’action transcriptionnelle du glucose, notre équipe a développé une technique d’ARN interférent (ARNi) permettant d’inhiber sélectivement l’expression de ChREBP. Dans des hépatocytes isolés de souris transfectés avec l’ARNi ChREBP, une forte concentration de glucose (25 mM) n’est plus capable de stimuler l’expression des gènes glycolytiques (L-PK) et lipogéniques (ACC, FAS) [16]. De plus, l’accumulation de lipides est fortement diminuée en l’absence de ChREBP dans ces cellules. Ces résultats indiquent que ChREBP est un déterminant essentiel de la synthèse de lipides in vitro. Par la suite, une analyse par immunoprécipitation de la chromatine a permis de montrer que ChREBP agit de façon directe en interagissant avec les promoteurs de ces gènes [17], en formant un hétérotétramère avec son partenaire protéique, le facteur de transcription Max-like protein X (Mlx) [18].
Régulation de l’activité de ChREBP
Le mécanisme par lequel ChREBP est activé par le glucose n’est pas totalement élucidé. Le groupe de Uyeda a montré, en construisant une protéine de fusion ChREBP-GFP (green fluorescent protein) que ChREBP est rapidement relocalisé du cytoplasme vers le noyau en réponse à de fortes concentrations de glucose [19]. Notre équipe a confirmé ces résultats in vivo sur la protéine ChREBP endogène, dans le foie de souris soumises à un régime riche en hydrates de carbone, grâce à une technique de séparation des protéines cytoplasmiques et nucléaires [20]. Le modèle actuellement proposé pour l’activation de ChREBP est le suivant : lorsque les concentrations de glucose sont faibles, ChREBP est localisé dans le cytoplasme et phosphorylé sur la sérine 196 (Sér196), adjacente au domaine NLS, et sur la thréonine 666 (Thr666) localisée dans le motif bHLH-LZ. Ces deux résidus sont des sites potentiels de phosphorylation par la protéine kinase A (PKA) [21]. Lorsque les concentrations de glucose augmentent, le flux de glucose entraîne la formation de xylulose 5-phosphate (Xu5P), un intermédiaire de la voie des pentoses-phosphate. Le Xu5P est capable d’activer la protéine phosphatase 2A (PP2A) qui déphosphoryle ChREBP sur la Sér196 et entraîne sa translocation dans le noyau. La PP2A déphosphoryle ensuite ChREBP sur la Thr666 pour lui permettre d’interagir avec les séquences ChoRE de ses gènes cibles et d’induire leur transcription [21] (Figure 3). Le développement récent au laboratoire d’un anticorps spécifique reconnaissant la forme phosphorylée de ChREBP sur la Sér196 nous a permis de déterminer l’état de phosphorylation de ChREBP endogène. Nous avons ainsi pu montrer qu’une injection de glucagon (qui active la PKA) dans le foie de souris réalimentées avec un régime hyperglucidique, où ChREBP est nucléaire, entraînait sa phosphorylation sur la Sér196 ainsi que sa relocalisation au cytosol. Ces résultats revèlent pour la première fois l’importance de cette phosphorylation in vivo [22].
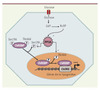 |
Figure 3. Régulation de l’activité transativatrice de ChREBP. La régulation de l’activité transactivatrice de ChREBP nécessite la mise en place d’un mécanisme de phosphorylation/déphosphorylation contrôlant sa localisation cellulaire ainsi que son activité de liaison à l’ADN. Ainsi, selon le modèle actuellement proposé, en présence de faibles concentrations de glucose, ChREBP serait présent inactif dans le cytosol sous forme phosphorylée sur la Sér196 et la Thr666. La translocation de ChREBP dans le noyau et sa capacité de fixation sur l’ADN en réponse à des fortes concentrations de glucose semblent être contrôlées par un mécanisme de déphosphorylation transmise par la phosphatase 2A (PP2A), activée spécifiquement par le xylulose 5-phosphate (Xu5P). G6P : glucose 6-phosphate. |
Implication de ChREBP dans le développement de la résistance à l’insuline et de la stéatose hépatique
Peu d’études se sont intéressées à la fonction de ChREBP in vivo. En 2004, le groupe de Uyeda a généré des souris invalidées pour le gène ChREBP [23]. Dans le foie de ces souris, l’expression des gènes de la glycolyse (L-PK) et de la lipogenèse (ACC, FAS) est diminuée. Par conséquent, le foie de ces souris contient moins de triglycérides, ce qui confirme l’importance de ChREBP dans le contrôle de la synthèse hépatique de lipides in vivo. Ces travaux ont également permis de montrer pour la première fois l’implication de ChREBP dans le contrôle de l’homéostasie glucidique. En effet, les souris déficientes en protéine ChREBP développent une intolérance au glucose et une résistance à l’insuline.
ChREBP et stéatose hépatique : le modèle des souris ob/ob
Bien qu’elle soit fortement exprimée dans le foie, ChREBP est également exprimée dans d’autres tissus impliqués dans l’homéostasie glucidique tels que le tissu adipeux, le muscle, le pancréas ou encore le cerveau. De ce fait, l’interprétation de l’invalidation globale de ChREBP chez la souris s’avère complexe. Afin d’étudier les conséquences de l’invalidation de ChREBP spécifiquement dans le foie, notre groupe a développé une approche d’ARNi véhiculé par un adénovirus dans un contexte physiopathologique de stéatose hépatique, celui des souris ob/ob. Ces souris sont déficientes pour le gène codant la leptine, une hormone synthétisée par le tissu adipeux qui agit au niveau central pour inhiber la prise alimentaire et augmenter la dépense énergétique [24]. Les souris ob/ob sont hyperphagiques, inactives et deviennent obèses. Elles sont également résistantes à l’insuline et développent une stéatose hépatique due en partie à une glycolyse et une lipogenèse exacerbées [25].
Afin d’établir un lien entre le facteur de transcription ChREBP et la stéatose hépatique des souris ob/ob, nous avons tout d’abord montré que l’expression génique de ChREBP est fortement augmentée dans le foie de ces souris [26]. De façon intéressante, au cours du jeûne, la forme nucléaire de ChREBP est présente chez les souris ob/ob, alors qu’elle est indétectable chez les souris contrôles, ce qui suggère que ChREBP pourrait être responsable des taux importants de lipogenèse mesurés au cours du jeûne chez ces animaux [26]. Dans le but de déterminer si la diminution de l’expression de ChREBP pourrait avoir des effets bénéfiques sur le phénotype des souris ob/ob, un adénovirus exprimant un ARNi de ChREBP a été injecté à ces animaux. Deux jours après l’injection de l’adénovirus, la diminution de l’expression hépatique de ChREBP est de 60 % et persiste après 7 jours. L’atténuation de l’expression de ChREBP s’accompagne d’une diminution des taux de lipogenèse et de la quantité de lipides dans le foie. Comme attendu, l’expression des gènes de la L-PK, de l’ACC et de la FAS est diminuée. De façon intéressante, l’inhibition de ChREBP affecte également l’expression de la stéaroyl-CoA désaturase 1 (SCD-1), l’enzyme qui catalyse la biosynthèse des acides gras mono-insaturés, et celle de la glycéraldéhyde 3-phosphate acyltransférase (GPAT), l’enzyme qui contrôle la première étape de la synthèse des triglycérides (Figure 1). Or, des travaux antérieurs ont mis en évidence un lien entre ces enzymes et la stéatose hépatique. En effet, l’invalidation de SCD-1 [27] ou de GPAT [28] chez les souris ob/ob diminue l’accumulation hépatique de lipides et améliore le phénotype des animaux. L’ensemble de ces résultats montrent que ChREBP, en contrôlant de façon coordonnée l’expression des gènes de la glycolyse, de la lipogenèse et de la synthèse des triglycérides, est un régulateur central des quantités d’acides gras dans le foie.
Résistance à l’insuline et stéatose hépatique
Bien que de nombreux travaux, aussi bien chez l’homme que chez l’animal, aient établi une association entre résistance à l’insuline et stéatose hépatique, les mécanismes permettant d’expliquer ce lien ne sont pas clairement établis. Une étude récente de l’équipe de Shulman suggère que la stéatose hépatique induite par la surexpression de la GPAT1 dans le foie serait suffisante pour conduire au développement d’une résistance à l’insuline [29]. En accord avec ce concept, la diminution de la stéatose consécutive à l’inhibition de l’expression de ChREBP chez les souris ob/ob s’accompagne d’une restauration de la sensibilité hépatique à l’insuline. En effet, alors que la phosphorylation par l’insuline de la protéine kinase B (Akt/PKB) et des kinases ERK1 et ERK2 (extracellular signal-regulated kinases 1 et 2), acteurs moléculaires majeurs de la voie de signalisation insulinique [30], est diminuée chez les souris ob/ob, l’inhibition de ChREBP permet de restaurer cette phosphorylation, témoin d’une amélioration de la sensibilité à l’insuline dans le foie des souris ob/ob traitées [26].
Dans un contexte physiologique, l’une des principales actions de l’insuline dans le foie est d’inhiber l’expression des enzymes de la néoglucogenèse responsables de la production hépatique de glucose en période de jeûne. Dans des conditions de résistance à l’insuline, ce frein est levé et la production hépatique de glucose participe à l’installation de l’hyperglycémie. Chez les souris ob/ob traitées avec l’adénovirus portant l’ARNi de ChREBP, l’amélioration de la voie de signalisation de l’insuline dans le foie s’accompagne d’une diminution de l’expression des enzymes clés de la néoglugogenèse et contribue ainsi à diminuer la glycémie des souris [26].
De façon intéressante, la sensibilité à l’insuline est restaurée non seulement dans le foie mais également dans le muscle squelettique et le tissu adipeux, dans lesquels la phosphorylation d’Akt en réponse à l’insuline est significativement augmentée. Puisque l’atténuation de l’expression de ChREBP touche le foie de façon spécifique, les conséquences métaboliques observées dans le muscle et le tissu adipeux sont indirectes et s’expliquent probablement par une amélioration de la lipidémie, de la glycémie et de l’insulinémie chez ces souris.
Bien que la lipogenèse de novo participe à l’accumulation de triglycérides dans le foie des patients NAFLD, l’implication de ChREBP n’a pas été démontrée chez l’Homme. Il est cependant intéressant de noter que le gène codant ChREBP a été identifié comme appartenant à la région chromosomique délétée dans le syndrome de Williams-Beuren, une maladie neurodéveloppementale (WBSCR14) [14, 31] dont environ 75 % des patients présentent une altération de leur tolérance au glucose ou un diabète silencieux qui pourrait s’expliquer par l’absence de ChREBP [32].
Conclusion
La découverte récente du facteur de transcription ChREBP a permis une avancée importante dans la compréhension du contrôle du métabolisme du glucose. ChREBP apparaît comme un régulateur central de la conversion du glucose alimentaire en lipides dans le foie. L’atténuation de son expression améliore le phénotype des souris ob/ob, ce qui suggère qu’une approche thérapeutique pour diminuer l’activité de ChREBP pourrait avoir des effets bénéfiques dans le traitement des maladies métaboliques associées à la résistance à l’insuline et la stéatose hépatique.
Remerciements
Les auteurs tiennent à remercier Renaud Dentin, Fadila Benhamed, Pierre-Damien Denechaud, Véronique Fauveau, Fabienne Foufelle, Isabelle Hainault et Pascal Ferré pour leur contribution dans la réalisation de ce travail. Le soutien financier a été apporté par l’Agence Nationale pour la Recherche (ANR-05 PCOD-035-02), le Programme National de Recherche sur le Diabète (PNRD-2005) et l’ALFEDIAM.
Références
- Junquero D, Rival Y. Syndrome métabolique : quelle définition pour quel(s) traitement(s) ? Med Sci (Paris) 2005; 21 : 1045–53. [Google Scholar]
- Day CP, James OF. Hepatic steatosis: innocent bystander or guilty party ? Hepatology 1998; 27 : 1463–6. [Google Scholar]
- Mendez-Sanchez N, Arrese M, Zamora-Valdes D, Uribe M. Current concepts in the pathogenesis of nonalcoholic fatty liver disease. Liver Int 2007; 27 : 423–33. [Google Scholar]
- Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab 2003; 29 : 478–85. [Google Scholar]
- Nakamuta M, Kohjima M, Morizono S, et al. Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med 2005; 16 : 631–5. [Google Scholar]
- Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005; 115 : 1343–51. [Google Scholar]
- Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest 2008; 118 : 829–38. [Google Scholar]
- Foufelle F, Hegarty B, Bobard A, et al. Un nouveau rôle de l’insuline dans la régulation du métabolisme glucido-lipidique hépatique. Med Sci (Paris) 2005; 21 : 569–71. [Google Scholar]
- Shimano H, Horton JD, Shimomura I, et al. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest 1997; 99 : 846–54. [Google Scholar]
- Bécard D, Hainault I, Azzout-Marniche D, et al. Adenovirus-mediated overexpression of sterol regulatory element binding protein-1c mimics insulin effects on hepatic gene expression and glucose homeostasis in diabetic mice. Diabetes 2001; 50 : 2425–30. [Google Scholar]
- Liang G, Yang J, Horton JD, et al. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem 2002; 277 : 9520–8. [Google Scholar]
- Decaux JF, Antoine B, Kahn A. Regulation of the expression of the L-type pyruvate kinase gene in adult rat hepatocytes in primary culture. J Biol Chem 1989; 264 : 11584–90. [Google Scholar]
- Stoeckman AK, Towle HC. The role of SREBP-1c in nutritional regulation of lipogenic enzyme gene expression. J Biol Chem 2002; 277 : 27029–35. [Google Scholar]
- Yamashita H, Takenoshita M, Sakurai M, et al. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci USA 2001; 98 : 9116–21. [Google Scholar]
- Cairo F, Rotundo R, Frazzingaro G, et al. Diabetes mellitus as a risk factor for periodontitis. Minerva Stomatol 2001; 50 : 321–30. [Google Scholar]
- Dentin R, Pegorier JP, Benhamed F, et al. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem 2004; 279 : 20314–26. [Google Scholar]
- Ishii S, Iizuka K, Miller BC, Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci USA 2004; 101 : 15597–602. [Google Scholar]
- Ma L, Sham YY, Walters KJ, Towle HC. A critical role for the loop region of the basic helix-loop-helix/leucine zipper protein Mlx in DNA binding and glucose-regulated transcription. Nucleic Acids Res 2007; 35 : 35–44. [Google Scholar]
- Kawaguchi T, Takenoshita M, Kabashima T, Uyeda K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc Natl Acad Sci USA 2001; 98 : 13710–5. [Google Scholar]
- Dentin R, Benhamed F, Pegorier JP, et al. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J Clin Invest 2005; 115 : 2843–54. [Google Scholar]
- Kabashima T, Kawaguchi T, Wadzinski BE, Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003; 100 : 5107–12. [Google Scholar]
- Denechaud PD, Bossard P, Lobaccaro JM, et al. ChREBP, but not LXRs, is required for the induction of glucose-regulated genes in mouse liver. J Clin Invest 2008; 118 : 956–64. [Google Scholar]
- Iizuka K, Bruick R K, Liang G, et al. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci USA 2004; 101 : 7281–6. [Google Scholar]
- Campfield LA, Smith FJ, Burn P. The OB protein (leptin) pathway: a link between adipose tissue mass and central neural networks. Horm Metab Res 1996; 28 : 619–32. [Google Scholar]
- Lindstrom P. The physiology of obese-hyperglycemic mice (ob/ob mice). Scientific World J 2007; 7 : 666–85. [Google Scholar]
- Dentin R, Benhamed F, Hainault I, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006; 55 : 2159–70. [Google Scholar]
- Cohen P, Miyazaki M, Socci ND, et al. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science 2002; 297 : 240–3. [Google Scholar]
- Xu H, Wilcox D, Nguyen P, et al. Hepatic knockdown of mitochondrial GPAT1 in ob/ob mice improves metabolic profile. Biochem Biophys Res Commun 2006; 349 : 439–48. [Google Scholar]
- Nagle CA, An J, Shiota M, et al. Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase 1 in rats causes insulin resistance. J Biol Chem 2007; 282 : 14807–15. [Google Scholar]
- Capeau J. Voies de signalisation de l’insuline : mécanismes affectés dans l’insulino-résistance. Med Sci (Paris) 2003; 19 : 834–9. [Google Scholar]
- De Luis O, Valero MC, Jurado LA. WBSCR14, a putative transcription factor gene deleted in Williams-Beuren syndrome: complete characterisation of the human gene and the mouse ortholog. Eur J Hum Genet 2000; 8 : 215–22. [Google Scholar]
- Cherniske E M, Carpenter T O, Klaiman C, et al. Multisystem study of 20 older adults with Williams syndrome. Am J Med Genet 2004; 131 : 255–64. [Google Scholar]
Liste des figures
|
Figure 1. Étapes métaboliques conduisant au développement de la stéatose hépatique. Lors du développement de la stéatose hépatique, les lipides qui s’accumulent dans le foie sont majoritairement des triglycérides (TG). En raison de l’état de résistance à l’insuline caractéristique du patient obèse pré-diabétique, la lipolyse au niveau du tissu adipeux n’est plus efficacement inhibée par l’insuline et des concentrations importantes d’acides gras non estérifiés (AGNE) sont libérées dans la circulation sanguine. Ces acides gras (AG) sont captés par le foie. En parallèle, et de manière paradoxale, la synthèse de novo d’acides gras par la voie de la lipogenèse est stimulée par l’hyperglycémie et l’hyperinsulinémie. La combinaison de l’augmentation de la voie de la lipogenèse et de la diminution de la voie de β-oxydation des acides gras conduit au développement de la stéatose hépatique. Les TG hépatiques sont stockés dans des gouttelettes lipidiques ou sécrétés dans le sang sous forme de lipoprotéines de très faible densité (VLDL). L’accumulation excessive des concentrations de TG hépatiques exacerbe le phénotype de résistance à l’insuline dans le foie, conduisant à l’augmentation de la production hépatique de glucose. Au niveau du muscle squelettique, les concentrations élevées d’AGNE conduisent à une résistance à l’insuline et à une diminution de l’utilisation de glucose dans ce tissu. |
| Dans le texte | |
|
Figure 2. Contrôle transcriptionnel des voies de la glycolyse et de la lipogenèse. En réponse à une ingestion d’hydrates de carbone, l’augmentation de la glycémie stimule la sécrétion d’insuline par les cellules β du pancréas endocrine. Dans le foie, l’insuline active l’expression des gènes de la glycolyse et de la lipogenèse, nécessaires à la synthèse de lipides. L’effet transcriptionnel de l’insuline est transmis par le facteur de transcription SREBP-1c qui stimule la glucokinase (GK), les enzymes de la lipogenèse [l’acétyl-CoA carboxylase (ACC), la fatty acid synthase (FAS)], ainsi que la stéaroyl-CoA désaturase 1 (SCD-1) qui catalyse la synthèse d’acides gras mono-insaturés et la glycéraldéhyde 3-phosphate acyltransférase (GPAT) qui permet la synthèse de triglycérides (TG). L’action du glucose est quant à elle relayée par le facteur de transcription ChREBP, qui, induit par le xylulose 5-phosphate (Xu5P), un intermédiaire métabolique de la voie des pentoses, stimule l’expression de la L-pyruvate kinase (L-PK) et agit en synergie avec SREBP-1c pour induire les gènes de la lipogenèse. G6P : glucose 6-phosphate ; PEP : phosphoénolpyruvate. |
| Dans le texte | |
|
Figure 3. Régulation de l’activité transativatrice de ChREBP. La régulation de l’activité transactivatrice de ChREBP nécessite la mise en place d’un mécanisme de phosphorylation/déphosphorylation contrôlant sa localisation cellulaire ainsi que son activité de liaison à l’ADN. Ainsi, selon le modèle actuellement proposé, en présence de faibles concentrations de glucose, ChREBP serait présent inactif dans le cytosol sous forme phosphorylée sur la Sér196 et la Thr666. La translocation de ChREBP dans le noyau et sa capacité de fixation sur l’ADN en réponse à des fortes concentrations de glucose semblent être contrôlées par un mécanisme de déphosphorylation transmise par la phosphatase 2A (PP2A), activée spécifiquement par le xylulose 5-phosphate (Xu5P). G6P : glucose 6-phosphate. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.