")
")
| Issue |
Med Sci (Paris)
Volume 22, Number 10, Octobre 2006
|
|
|---|---|---|
| Page(s) | 806 - 808 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20062210806 | |
| Published online | 15 October 2006 | |
Le chien et son génome
The dog and its genome
Laboratoire de Génétique et Développement, UMR 6061, CNRS-Université de Rennes 1, IFR 140, Génomique Fonctionnelle et Santé, 2, avenue Léon Bernard, 35043 Rennes Cedex, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Et de cinq !
Le 8 décembre dernier, la revue Nature publiait la séquence complète du génome du chien essentiellement réalisée par le BROAD Institute (Boston, MA, États-Unis) sous la direction de Kerstin Lindblad-Toh [1]. Ainsi, après ceux de l’homme, de la souris, du rat et du chimpanzé, le chien a été choisi pour compléter cette liste déjà impressionnante de génomes pour lesquels une connaissance très approfondie est maintenant disponible. Pourquoi le chien et pas un autre mammifère ? Quelles avancées cette séquence va-t-elle permettre ? Est-il nécessaire d’allonger encore cette liste ? Telles sont les questions auxquelles nous tenterons de répondre.
Mais d’abord, il convient de rappeler quelques faits sur la séquence proprement dite et son établissement. La séquence publiée par K. Lindblad-Toh et ses collaborateurs correspond à celle d’un chien unique, une femelle boxer, sélectionnée parmi beaucoup d’autres chiens de diverses races pour son faible niveau de polymorphisme. Ces deux éléments, associés à l’utilisation d’un programme d’assemblage amélioré et à l’utilisation de cartes génomiques denses et robustes expliquent pour beaucoup la qualité exceptionnelle de cette séquence, supérieure à celles des autres génomes, séquence humaine exceptée. Autre point intéressant à souligner, la séquence publiée résulte uniquement de l’assemblage d’un shotgun profond du génome canin entier, démontrant, s’il en était encore besoin, la puissance de cette approche pourtant décriée à l’excès lorsque fut proposée en 2002 son adaptation aux génomes de mammifères.
Le chien est la première espèce domestiquée par l’homme comme l’attestent les données archéologiques et de biologie moléculaire, les dates les plus communément avancées se situant entre – 13 000 et – 15 000 ans. Par ailleurs, les analyses d’ADN mitochondriaux et génomiques de dizaines d’échantillons de loups et de chiens de très nombreuses races indiquent clairement que toute la population canine actuelle dériverait d’une origine commune de loup asiatique, canis lupus [2] sans toutefois éliminer la possibilité de croisements occasionnels avec d’autres espèces du genre canis. Cette origine unique alliée à la très grande diversité anatomique, comportementale et de susceptibilité aux maladies constitue la base de l’intérêt du chien comme modèle à nul autre pareil pour l’analyse des relations génotypes/phénotypes.
Comme chacun peut le constater, si le Berger allemand ou le Husky ont conservé un aspect général peu éloigné du loup, comment s’imaginer de prime abord que le Chihuahua ou le Greyhound, pour ne citer que ces deux races, puissent être des représentants de la même espèce. Au cours des siècles, et singulièrement depuis 300 à 400 ans, l’homme a exercé une pression de sélection énorme en réalisant des croisements orientés vers la création de plus de 300 races ayant des phénotypes répondant à des besoins divers comme la chasse, le gardiennage de troupeaux ou d’installations, ou plus simplement de compagnie et d’aide à la personne [3, 4].
Sur le plan anatomique, ces croisements dirigés ont produit une variété que n’offre aucune autre espèce mammifère. Ils ont aussi modelé des aptitudes comportementales aussi diverses que celles exprimés par le Labrador ou le Pit-bull, par exemple, et une capacité au moins de certaines races à communiquer avec l’homme très supérieure à celle exprimée par le loup ou le chimpanzé [5]. Malheureusement cette sélection fondée sur des caractères phénotypiques ou comportementaux s’est accompagnée de la co-sélection d’allèles morbides responsables à l’état homozygote de nombreuses maladies génétiques de sorte que la plupart des races de chiens – chacune d’entre elles étant peu ou prou un véritable isolat génétique – souffrent d’un grand nombre de maladies souvent spécifiques de races ou de groupes de races apparentées. Au-delà des maladies génétiques à transmission mendélienne simple, beaucoup de races de chien présentent une susceptibilité exagérée à des maladies génétiques complexes, comme par exemple les cancers ou les maladies auto-immunes.
Cette situation malheureuse et préoccupante pour tous les amis des chiens, éleveurs, vétérinaires, cynophiles, est une opportunité pour la génétique médicale, d’autant que nombre de ces maladies sont communes à l’homme et au chien, que les connaissances médicales des affections du chien sont les plus développées du règne animal et que le chien, vivant au contact des hommes, est soumis aux mêmes conditions environnementales [6, 7]. Ces considérations ont fait prendre conscience de l’intérêt unique que le chien pouvait avoir comme modèle génétique particulièrement favorable à l’identification des allèles de gènes morbides. Cette prise de conscience s’est traduite par la construction depuis une dizaine d’années de nombreuses cartes génomiques [8, 9], d’un séquençage léger (1,5x) [10], et très récemment du séquençage complet de ce génome. Par ailleurs, on est parvenu, sur la base d’un million de lectures de séquences aléatoires réalisées à partir de 10 ADN génomiques provenant de races différentes, à l’identification d’un très grand nombre de sites nucléotidiques variants (SNP, single nucleotide polymorphism) [1].
Les premières analyses du polymorphisme génétique montrent déjà ce à quoi on pouvait s’attendre compte tenu de l’histoire des races : le déséquilibre de liaison que l’on peut mesurer par l’analyse de la distribution des microsatellites ou des SNP peut s’étendre sur des distances beaucoup plus grandes que dans le génome humain. Il en résulte que le nombre de sites polymorphes à analyser pour délimiter les régions génomiques impliquées dans la susceptibilité aux maladies complexes devrait être beaucoup plus restreint dans le modèle canin que chez l’homme [1, 11]. Si on ajoute à cela le fait que les problèmes éthiques, sans être nuls, sont beaucoup moins prégnants, et qu’il est possible de réaliser à la demande de nouveaux croisements, on comprend toute l’importance que peut revêtir le chien comme modèle pour l’analyse des causes des maladies génétiques à transmission mendélienne simple ou complexe.
Mais le chien est aussi un extraordinaire modèle pour l’analyse des relations génotype-phénotype et pour comprendre les modalités de la ségrégation de caractères anatomiques. Quels sont les gènes – et pour chacun quels allèles – contrôlent le développement du crâne, du nez, des pattes… ou cette extrême diversité de robe et de pelage pour ne citer que quelques traits phénotypiques ? Les fondements génétiques de la diversité comportementale et d’aptitude qui ségrègent dans les diverses races canines sont aussi un sujet d’interrogation qui pourra être abordé désormais par la délimitation des régions génomiques soumises à la pression de sélection exercée de façon particulière dans chaque race.
Pour tous ces sujets d’étude, les outils génomiques que représentent les milliers de SNP localisés dans le génome canin sont en place. Il convient maintenant d’analyser leur fréquence de distribution dans la population canine en général, et dans des cohortes de chiens choisis en fonction des problématiques à aborder. Compte tenu de l’hétérogénéité génétique de l’espèce prise dans son ensemble (qui est de même amplitude que celle observée chez l’homme) et de la grande homogénéité intra-race, avec des régions génomiques caractérisées par de grandes distances en déséquilibre de liaison (LD) au sein de zones de faibles LD, ces analyses seront à l’évidence beaucoup plus facilement abordables.
Comme nous l’avons indiqué pour commencer, le génome du chien est le cinquième génome mammifère entièrement séquencé. Peut-on, doit-on en séquencer d’autres ? En dehors des avantages propres apportés par le modèle canin, la détermination de la séquence complète de ce génome comme celle de tout autre génome de mammifères et de son annotation permet une réappréciation de l’annotation du génome humain. Depuis les premières estimations d’environ 100 000 gènes à celle actuellement retenue d’environ 25 000, le nombre de gènes identifiés dans le génome humain s’est considérablement réduit au cours des cinq dernières années. La difficulté d’identification précise des gènes tient pour beaucoup à leur structure morcelée de sorte que dans beaucoup de cas seule la comparaison des séquences d’autres génomes de mammifères permet de lever les ambiguïtés. La comparaison des séquences de génomes de mammifères permet aussi d’aborder la question des séquences conservées non géniques, mais importantes pour la structuration ou la régulation de l’expression génique. Enfin, la connaissance fine des génomes permet également d’aborder différemment les questions d’évolution et de spéciation [12]. Tout ceci explique que plusieurs génomes de mammifères soient des candidats au séquençage (http://www.broad.mit.edu/mammals; http://www.genome.gov/10002154). Malheureusement, même si des progrès technologiques sont continuellement réalisés et abaissent régulièrement le coût du séquençage, celui-ci n’en reste pas moins très élevé (la détermination de la séquence du génome canin aura coûté 30 à 40 millions de dollars). Depuis peu, on s’est donc orienté vers des séquençages légers partiels capables d’identifier la très grande majorité des gènes, un grand nombre de sites polymorphes et de zones non géniques conservées. Malheureusement ces séquences partielles restent extrêmement fragmentées – plus d’un million de contigs après assemblage – et ne donnent donc pas de vision globale des génomes [10]. L’analyse comparative des séquences complètes disponibles montre que les génomes de mammifères ont évolué, notamment par des déplacements en bloc de pans entiers plus ou moins grands de chromosomes, appelés blocs de synténie, mais que dans ces blocs de synténie, quelle que soit leur taille, l’ordre des gènes reste singulièrement conservé (Figure 1). La construction de cartes génomiques denses réalisée par la méthode des hybrides d’irradiation et ordonnant des milliers de marqueurs de gènes permettrait de transformer pour un coût et un effort marginal l’information très fractionnée résultant d’un shotgun léger en une structure continue enchâssée dans le caryotype [9].
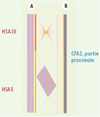 |
Figure 1. Diagramme comparant l’alignement des gènes déduit des séquences humaine (A) et canine (B). La partie proximale du chromosome 2 de chien (CFA2) correspond à 2 fragments synténiques provenant des chromosomes humains 5 et 10 (HSA5 et HSA10). Toutefois, pour rendre compte de l’ordre général des gènes correspondant à HSA5, le fragment synténique doit être scindé en deux parties ayant la même orientation mais des positions alternées dans CFA2. Enfin on peut noter qu’à l’intérieur des différents fragments pris deux à deux, l’ordre des gènes est parfaitement respecté (© Hitte et Derrien). |
Références
- Lindblad-Toh K, Wade CM, Mikkelsen TS, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005; 438 : 803–19. [Google Scholar]
- Ostrander EA, Wayne RK. The canine genome. Genome Res 2005; 15 :1706–16. [Google Scholar]
- Ostrander EA, Galibert F, Patterson DF. Canine genetics comes of age. Trends Genet 2000; 16 : 117–24. [Google Scholar]
- Parker HG, Kim LV, Sutter NB, et al. Genetic structure of the purebred domestic dog. Science 2004; 304 : 1160–4. [Google Scholar]
- Hare B, Brown M, Williamson C, Tomasello M. The domestication of social cognition in dogs. Science 2002; 298 : 1634–6. [Google Scholar]
- Sutter NB, Eberle MA, Parker HG, et al. Extensive and breed specific linkage disequilibrium in Canis familiaris. Genome Res 2004; 14 : 2388–96. [Google Scholar]
- Galibert F, André C, Hitte C. Le chien, un modèle pour la génétique des mammifères. Med Sci (Paris) 2004; 20 : 761–6. [Google Scholar]
- Breen M, Hitte C, Lorentzen TD, et al. An integrated 4249 marker FISH/RH map of the canine genome. BMC Genomics 2004; 5 : 1–11. [Google Scholar]
- Hitte C, Madeoy J, Kirkness EF, et al. Facilitating genome navigation: survey sequencing and dense radiation-hybrid gene mapping. Nat Rev Genet 2005; 6 : 643–8. [Google Scholar]
- Kirkness EF, Bafna V, Halpern AL, et al. The dog genome: survey sequencing and comparative analysis. Science 2003; 301 : 1898–903. [Google Scholar]
- Sutter NB, Ostrander EA. Dog star rising: the canine genetic system. Nat Rev Genet 2004; 5 : 900–10. [Google Scholar]
- Murphy WJ, Larkin DM, Everts-Van der Wind A, et al. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science 2005; 309 : 613–7. [Google Scholar]
© 2006 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Diagramme comparant l’alignement des gènes déduit des séquences humaine (A) et canine (B). La partie proximale du chromosome 2 de chien (CFA2) correspond à 2 fragments synténiques provenant des chromosomes humains 5 et 10 (HSA5 et HSA10). Toutefois, pour rendre compte de l’ordre général des gènes correspondant à HSA5, le fragment synténique doit être scindé en deux parties ayant la même orientation mais des positions alternées dans CFA2. Enfin on peut noter qu’à l’intérieur des différents fragments pris deux à deux, l’ordre des gènes est parfaitement respecté (© Hitte et Derrien). |
| Dans le texte | |
 |
Le chien et son génome, par Andrzej Krauze (© octobre 2006). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.