")
")
| Issue |
Med Sci (Paris)
Volume 41, Number 6-7, Juin-Juillet 2025
|
|
|---|---|---|
| Page(s) | 585 - 592 | |
| Section | Repères | |
| DOI | https://doi.org/10.1051/medsci/2025083 | |
| Published online | 7 juillet 2025 | |
Épidémiologie moléculaire par séquençage des acides nucléiques d’origine virale présents dans les eaux usées
Molecular epidemiology of viruses sequenced from wastewater
1
Université Côte d’Azur, CNRS UMR 7275, INSERM U1323, Institut de pharmacologie moléculaire et cellulaire, Sophia Antipolis, France
2
IHU Méditerranée infection, Aix-Marseille université, Marseille, France
3
IHU Respirera, Sophia Antipolis, France
4
3IA Côte d’Azur, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
L’analyse métagénomique des acides nucléiques présents dans les eaux usées est un outil épidémiologique prometteur pour suivre la propagation des virus dans de grands bassins démographiques. Son utilisation lors de la pandémie de COVID-19 a permis de surv eiller la circulation du SARS-CoV-2 sans nécessiter la collecte de multiples échantillons individuels. Cette approche permet de suivre non seulement les infections symptomatiques mais aussi les infections asymptomatiques. Initialement basée sur une détection par RT-qPCR, l’introduction du séquençage métagénomique améliore cette détection en fournissant des informations plus détaillées. L’expérience acquise avec la COVID-19 suggère d’étendre désormais cet outil épidémiologique puissant à d’autres virus également détectables dans les eaux usées. Dans cet article, nous décrivons les différentes méthodes mises en œuvre, et nous discutons les enjeux d’un déploiement rapide à une échelle internationale pour mieux appréhender la circulation des virus pathogènes à l’échelle mondiale.
Abstract
Virus surveillance using metagenomic analysis of sequences from wastewater appears to be a promising epidemiological tool for monitoring the spread of viruses in large populations. Its use during the COVID-19 pandemic enabled the monitoring of SARS-CoV-2 circulation without requiring the collection of multiple individual samples. This approach allows both symptomatic and asymptomatic infections to be monitored in a highly cost-effective way. Initially based on PCR detection, the introduction of nucleic acid sequencing has improved this tool by providing more detailed metagenomic information. Experience with COVID-19 pandemics suggests that this epidemiological tool should now be extended to other viruses detectable in wastewater. This review discusses the different methods used, highlighting the challenges of a rapid deployment on an international scale to better understand the global circulation of viral pathogens.
© 2025 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Pascal Barbry).
L’analyse des eaux usées est un outil puissant de surve illance e nvironnementale, capable d’identifier et de quantifier des risques infectieux potentiels pour la santé humaine. Elle a permis d’étudier la circulation de dizaines d’agents pathogènes présents dans les eaux usées [1], mais aussi l’émergence de bactéries résistantes aux antibiotiques, ou la présence de médicaments, de drogues et de molécules toxiques [2]. La détection de séquences d’acides nucléiques d’origine virale dans des préparations d’eaux usées permet d’évaluer leur concentration, et l’analyse des séquences détectées permet de préciser la présence d’éventuels variants. Cette approche métagénomique permet ainsi d’apprécier le niveau de circulation des agents infectieux et/ou le niveau d’exposition des populations (Figure 1).
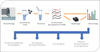 |
Figure 1 Schéma bilan illustrant les étapes du processus de surveillance de la circulation des séquences virales basé sur le séquençage des eaux usées. |
Dès le milieu du XIXe siècle, les travaux de John Snow1 avaient établi un lien entre le mode de transmission du choléra et la contamination des eaux. La démonstration par Paul, Trask et Culotta d’une transmission du poliovirus par les eaux usées conduisit Melnick à mettre en place une surveillance épidémiologique du poliovirus à partir de 1947 [3, 4]. Cette approche fut ensuite étendue à d’autres virus dans les années 1980 [5, 6]. Une identification directe des virus à partir de la détection de leurs acides nucléiques se mit alors progressivement en place, fournissant de nouvelles informations sur leurs niveaux de circulation dans la population et permettant d’alerter en cas d’émergence épidémique. Pendant la pandémie de COVID-19, de telles mesures réalisées par RT-qPCR (PCR quantitative après transcription inverse) ont ainsi permis de surveiller la propagation du SARS-CoV-2 dans la population générale et de prédire des remontées épidémiques avant que celles-ci ne soient observables cliniquement [7, 8]. Cette analyse épidémiologique indirecte est réalisable en temps quasi réel, et elle peut facilement s’étendre à tout virus excrété dans les urines ou les selles.
Un avantage majeur de cette approche est de fournir des indications sur les infections asymptomatiques et pré-symptomatiques sans devoir recourir à des mesures individuelles sur un grand nombre d’individus [9], ainsi que sur des réservoirs non humains [10]. Chaque mesure étant représentative d’une large population, le prix de revient de telles analyses est extrêmement bas. En revanche, l’analyse métagénomique ne fournit aucune information sur la contribution à l’échelle individuelle.
La plupart des approches actuelles se basent sur des séquences virales de référence à partir desquelles sont déduites les amorces utilisées pour détecter les agents pathogènes par RT-qPCR. Un séquençage de plusieurs amplicons répartis sur toute la séquence permet de documenter les différents variants. Un séquençage « shotgun »2 de toutes les séquences génomiques présentes est en principe possible, mais la prédominance de séquences d’origines bactériennes et eucaryotes limite la sensibilité de la mesure des virus [11, 12].
Les mesures s’opèrent sous de multiples contraintes : intrinsèques, comme le type de virus (à génome ADN ou ARN, enveloppé ou non) ; extrinsèques, comme la température de stockage, et l’impact des contaminants organiques ou microbiens. La standardisation des mesures et des analyses menées à partir des eaux usées est essentielle, depuis la collecte jusqu’à l’analyse des données. Le présent article fait un état de l’art des protocoles développés, compare l’efficacité et les limites de différents protocoles d’échantillonnage et de purification des virus, d’extraction des acides nucléiques, de préparation des banques et d’analyse bio-informatique (Figure 2).
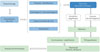 |
Figure 2 Détails des étapes du processus de surveillance de la circulation des séquences virales basé sur le séquençage des eaux usées. |
Méthodes d’échantillonnage, d’enrichissement et d’analyse
Une recherche bibliographique sur PubMed a identifié les articles utilisant la détection dans les eaux usées, par PCR ou RT-qPCR ou séquenç age, du SARS-CoV2, des coronavirus saisonniers, des rhinovirus, du virus respiratoire syncytial, des virus de la grippe, des virus des hépatites A et E, du virus mpox, ainsi que d’autres virus responsables d’infections respiratoires et gastro-intestinales (Tableau I).
Sélection d’études utilisant le séquençage d’acides nucléiques viraux sur des échantillons d’eaux usées. Entérovirus D68 (EVD-68), norovirus groupe II (NoVGII), adénovirus humain 41 (HAdV41), Hépatite A (HAV), virus de la rougeole (MeV), virus de la grippe A (IAV).
Echantillonnage et prélèvement
Les extraits obtenus à partir des eaux usées diffèrent de ceux obtenus à partir des échantillons cliniques du fait de la présence de multiples virus, à des con centrations variables, provenant d’un grand nombre d’individus. Leurs interactions avec les matières en suspension dans les eaux usées, et la présence possible de contaminants, tels que l’acide humique, interfèrent directement avec les mesures génomiques. Les caractéristiques propres des stations d’épuration, la densité des populations desservies, mais aussi la pluviométrie, la température, et la présence de certaines molécules chimiques peuvent aussi causer des différences compositionnelles importantes [13], imposant une normalisation.
La fréquence d’échantillonnage doit être adaptée à la taille et au taux de renouvellement du bassin de collecte, qui dépend notamment des niveaux de précipitations, et de la configuration géographique. Un pompage en continu sur 24 heures paraît plus adapté qu’un prélèvement ponctuel pour fournir des informations représentatives, reproductibles et avec un niveau de sensibilité élevé, même lorsque les niveaux de circulation viraux sont faibles [14]. Une fréquence hebdomadaire de prélèvement est habituellement suffisante, mais peut être augmentée en cas d’émergence épidémique [13]. Des volumes suffisants de collecte (40-1 000 mL) sont indispensables pour permettre la détection de virus faiblement abondants ou la présence de variants minoritaires. En revanche, l’augmentation des volumes analysés, de même que l’utilisation de matières plus concentrées mais plus complexes comme des boues, augmentent aussi la concentration des inhibiteurs enzymatiques et peuvent détériorer la qualité des analyses [15].
Préparation des acides nucléiques
Toutes les manipulations effectuées ont lieu dans des espaces confinés, afin d’assurer la sécurité des manipulateurs et empêcher toute dissémination. La concentration du SARS-CoV-2 dans les fèces de patients infectés peut atteindre 107 copies/mL. Elle peut descendre à moins de 103 copies/mL dans les eaux usées non traitées [16]. Moins de 1 % des séquences obtenues par séquençage non ciblé sont d’origine virale, avec une forte représentation de bactériophages et de virus environnementaux [12]. L’enrichissement des échantillons par des méthodes basées sur la taille, ou par précipitation différentielle est donc primordial. La purification des virus non enveloppés (tels que les rotavirus, ou les virus de l’hépatite A et E), plus résistants, est déjà bien décrite [17]. La purification des virus enveloppés (tels que le SARS-CoV-2), plus fragiles, s’est notablement améliorée avec la pandémie du COVID-19 [15].
Les acides nucléiques sont purifiés à partir d’extraits concentrés. Une ultrafiltration tangentielle permet de concentrer les virus [15]. Les complexes de virus enveloppés et de matières en suspension peuvent par exemple être récupérés par centrifugation (30 minutes x 10,000 g) [18]. Des ultracentrifugat ions à plus de 100,000 g pendant plusieurs heures permettent de culotter tous les virus, mais l’accès limité à des équipements d’ultracentrifugation restreint leur utilisation [19–21]. La procédure « Viradel » d’adsorptionélution des virus fonctionne bien pour les virus non enveloppés, plus résistants aux pH acides [15, 22, 23]. La protonation des protéines des capsides virales à pH acide facilite leur adsorption sur membrane électronégative, particulièrement en présence d’AlCl3 ou de MgCl2 [23, 24]. Après filtration de grands volumes (> 1 litre) sur une membrane, les virus sont élués en solution alcaline [7, 15], ou après adsorption sur membrane électropositive [15, 24]. Les variations des points isoélectriques entre virus, voire entre souches, peuvent nécessiter certaines adaptations [24, 25]. La précipitation au polyéthylène glycol (PEG) permet de purifier le SARS-CoV-2, le monkeypox virus, et le norovirus [22, 26–29], qui sont alors concentrées par filtration ou centrifugation [30]. Les particules virales peuvent aussi être complexées à de l’hydroxyde d’aluminium, pour être ensuite collectées par centrifugation en un temps de préparation réduit [12, 29, 31]. La floculation organique a été très utilisée avec les virus non enveloppés (norovirus, adénovirus et polyomavirus) [15]. Une alternative consiste à adsorber directement les acides nucléiques sur des billes de silice dans un milieu de forte concentration ionique, et d’extraire directement les acides nucléiques des eaux usées, sans nécessiter d’équipements, tout en restant compatible avec une analyse par séquençage [15].
Le choix des méthodes dépend de la nature des échantillons, des virus recherchés, et de l’équipement disponible dans le laboratoire. Dans tous les cas, il s’agit de diminuer au maximum la concentration en inhibiteurs de l’étape de PCR [7]. De nombreux kits commerciaux d’extraction des acides nucléiques ont été spécialement adaptés au traitement des eaux usées.
Détection de virus d’intérêt
Détection et quantification par PCR
La quantification par PCR en temps réel (qPCR), ou par PCR digitale (ddPCR : digital droplet PCR) utilise des amorces qui ne sont pas encore systématiquement standardisées (i.e. les gènes N, E, S pour SARS-CoV-2 ; les gènes C22L, F3L, F8L pour le virus mpox). Les eaux usées constituant un mélange complexe, une normalisation des extraits viraux en fonction de multiples paramètres décrits est essentielle, par exemple en calculant le rapport entre le virus d’intérêt et un indicateur présent en abondance et de niveau stable dans les eaux usées. De différences importantes sont toutefois observées en fonction du normalisateur choisi [32]. Le virus de la marbrure légère du piment (PMMoV), la créatinine (CRE), l’acide 5-hydroxyindoleacétique (5-HIAA), la caféine et son métabolite paraxanthine sont autant de biomarqueurs possibles [33]. Certains paramètres hydrochimiques tels que l’ammonium (NH4+) [34] ou d’indicateurs fécaux comme E.coli [15, 31, 35] ont aussi été utilisés. Cependant, l’absence de standardisation sur l’utilisation des biomarqueurs rend difficile la comparaison des études.
Génotypage
Un génotypage peut être réalisé par séquençage ciblé d’une zone critique amplifiée par PCR ou par la production de multiples amplicons chevauchants, répartis sur tout le génome. Un alignement de l’ensemble des séquences obtenues sur une référence connue permet ensuite de suivre l’émergence éventuelle d’un variant à partir d’une mesure de la fréquence de mutations [36].
Détection par séquençage
Les plateformes de séquençage de type Illumina génèrent des séquences de moins de 300 bases. Même si les séquences génomiques présentes sont déjà fragmentées avant séquençage, des lectures plus longues produites avec la technologie Oxford Nanopore, permettent d’améliorer la couverture, avec une précision actuellement supérieure à 99,49 % pour des flowcells Nanopore (contre 99,98 % sur Illumina) [37]. Différents outils bio-informatiques d’analyse comme Freyja [38], LoFreq [39] ou iVar [40] permettent d’établir la fréquence des différentes mutations, indépendamment de la technologie de séquençage employée.
L’identification des virus repose sur l’alignement des séquences générées (reads) avec une base de données de référence des virus d’intérêt. L’utilisation combinée de la qPCR ou ddPCR (PCR digitale) [41] (→) pour la quantification et du séquençage pour la détection et l’identification des nouveaux variants permet une description complète des échantillons [27].
(→) Voir m/s n° 2, 2015, page 180
Enrichissement et séquençage
Pré-amplification par PCR
La production de multiples amplicons chevauchants, répartis sur tout le génome, permet d'acquérir des informations sur l’ensemble du génome. Les dizaines d’amplicons obtenus fournissent une couverture génomique homogène sur tout le génome, avec une profondeur suffisante pour discriminer les différents variants [12, 42]. Plusieurs jeux d’amorces spécifiques ont été utilisés pour le SARS-CoV-2 [12].
Enrichissement par capture après hybridation
Plusieurs industriels proposent des kits d’enrichissement par capture des régions ciblées par hybridation. L’enrichissement permet d’identifier de nombreux virus simultanément dans les eaux usées, avec une couverture correcte des génomes d’intérêt [21]. Cela a été notamment appliqué pour les polyomavirus, les astrovirus et les norovirus [43]. L’enrichissement par capture d’hybrides constitue une méthode robuste pour le suivi épidémiologique simultané d’un large panel de virus.
Dans un séquençage métagénomique non ciblé, les séquences virales représentent moins d’1 % de tous les reads [37]. La sensibilité est alors le plus souvent insuffisante pour permettre la détection des variants viraux, même avec une forte profondeur de séquençage.
Analyse bio-informatique
Les pipelines d’analyse automatisés développés par le consortium ARTIC (https://artic.network/) ou par Oxford Nanopore (EPI2ME labs, https://github.com/epi2me-labs) sont utilisables pour la classification sous forme de scripts qui automatisent l’identification des séquences et des variants sur des serveurs distants. ARTIC et EPI2ME, tout comme la plupart des outils existants traitent des données issues de divers types de séquenceurs. Ils reposent sur des ressources nucléotidiques comme celles du NCBI (GenBank), ou de GISAID (global initiative on sharing all influenza data).
Applications et perspectives
Actuellement, la surveillance des eaux usées permet non seulement la détection de virus à tropisme intestinal, responsables des gastro-entérites [12, 20, 42, 43], mais aussi de certains virus respiratoires tels que le SARS-CoV-2, les virus de la grippe, le virus respiratoire syncytial et les coronavirus humains. La qPCR et la ddPCR [41] sont utilisées pour déterminer la concentration virale. Des séquençages permettent ensuite de détecter les variants circulants [12, 13, 15, 19, 23, 26–29, 31, 35, 37, 42–44]. L’approche a récemment été appliquée aux adenovirus, aux polyomavirus mais aussi au suivi de l’épidémie du virus mpox en 2022 [22, 42, 43].
Corrélation entre données cliniques et eaux usées
La concentration virale dans les eaux usées est très bien corrélée à l’incidence des cas dans la population générale. Ceci a été démontré dès 2013 avec la surveillance du poliovirus [45]. Plus récemment, plusieurs études ont montré que les concentrations des virus de la grippe A, du SARS-CoV-2, ou encore du virus respiratoire syncitial (VRS) dans les eaux usées étaient directement liées à l’incidence observée dans la population [7, 21, 31, 46].
La concentration virale dans les eaux usées précédant l’augmentation de l’incidence dans la population, peut servir d’indicateur précoce comme démontré en 2014 pour les norovirus et le virus de l’hépatite A [47].
Pour le SARS-CoV2, l’augmentation des concentrations dans les eaux usées précède d’une semaine l’augmentation de l’incidence des cas cliniques [28].
L’analyse des données sur les eaux usées présente l’avantage de donner une image globale de l’ensemble des cas d’une population, y compris les cas pré-symptomatiques et asymptomatiques.
Génotypage des variants des virus
La surveillance des virus dans les eaux usées par séquençage assure la possibilité de suivre les variants viraux en circulation. La clé de ce suivi repose sur une couverture suffis ante des régions critiques et sur un échantillonnage suffisant du matériel génétique [42].
Cette approche permet aussi la détection précoce de variants émergents. En septembre 2023, la détection de 825 reads sur un total de 361 675, soit 0,2 % de toutes les séquences spécifiques du variant BA.2.86 (Pirola) du SARS-CoV2 dans les eaux usées de la région de Nice (Alpes Maritimes), a permis de caractériser sa présence alors qu’aucun cas n’avait encore été rapporté à l’hôpital (Figure 3).
 |
Figure 3 Première identification du variant BA.2.86 (Pirola) par analyse des eaux usées de Nice en septembre 2023, effectué à partir d’un prélèvement de la station centrale d’épuration de la Métropole Nice Côte d’Azur, qui collecte les eaux usées d’environ 400 000 habitants (non publié, source IPMC). |
Étude des interactions potentielles des communautés virales
Le séquençage des eaux usées permet d’analyser globalement l’évolution des communautés virales et de décrire leur saisonnalité [42, 43]. En séquençant 1 408 échantillons composites, iss us de 12 points de collecte différents, Smith et al. ont montré les variations de communautés virales des eaux usées en fonction des saisons et des zones de collecte [43]. L’existence d’un effet de saisonnalité est en accord avec les observations cliniques faites pour certains virus intestinaux et respiratoires. Les virus respiratoires syncytiaux, les virus parainfluenza, les norovirus et les astrovirus ont été détectés plus fréquemment en automne, en hiver et au début du printemps, c’est-à-dire au même moment que les épidémies saisonnières. A contrario, quelques virus comme le coronavirus humain hCoV-229E montrent un décalage entre leur concentration virale dans les eaux usées et le nombre de cas rapportés. Ce type d’études peut également permettre de mettre en évidence certaines interactions entre espèces [43], contribuant ainsi à fournir une image plus précise de la circulation virale entre différentes populations hôtes.
Du point de vue épidémiologique, la surveillance des virus dans les eaux usées par séquençage constitue un outil très important. En améliorant notre connaissance sur les multiples voies de transmission des virus, de nouveaux champs d’études sur les mécanismes de compétition ou de symbiose entre variants et entre espèces sont en train de s’ouvrir. L’application d’approches similaires aux eaux noires des avions a été proposée pour cartographier la propagation mondiale des virus lors des vols internationaux [48]. Même si l’organisation du transport aérien rend difficile un tel suivi en ce moment, quelques expérimentations en vie réelle ont exploré une telle faisabilité [49]. Au-delà des seules approches génomiques, un dernier enjeu important est constitué par le développement d’outils complémentaires pour surveiller la dissémination des antibiotiques, des médicaments antiviraux, ou la présence de polluants et de molécules toxiques. Le suivi global de tous ces marqueurs et leur intégration permettra certainement d’améliorer significativement le suivi de l’état général de grandes populations.
Le succès de l’utilisation du séquençage dans la surveillance des virus des eaux usées repose désormais sur un déploiement approprié, y compris dans des régions confrontées à de mauvaises conditions sanitaires. Cette approche pourrait alors contribuer à détecter plus précocement de futurs risques sanitaires et à empêcher le déclenchement de nouvelles épidémies.
John Snow (1813-1858) est un médecin britannique, pionnier dans les domaines de l’anesthésie, de l’hygiène et la santé publique (ndlr).
En génomique, le séquençage « shotgun », littéralement « fusil de chasse » est une méthode utilisée pour séquencer des brins d’ADN aléatoires.
Remerciements
Ce travail a été soutenu par des financements gérés par l’Agence nationale de la recherche dans le cadre du programme France 2030 (Respirera : ANR-23-IAHU-0007, 4D-OMICs : ANR-21-ESRE-0052, 3IA : ANR-19-P3IA-0002; Sisp&Eau: ANRS-23-PEPR-MIE-0003; France Génomique: ANR-10-INBS-09-03 ; OBEPINE+ : ANR-24-MIEM-0004), ainsi que par le Conseil départemental 06 (2016-294DGADSH-CV), et la métropole Nice Côte d’Azur.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Kilaru P, Hill D, Anderson K, et al. Wastewater Surveillance for Infectious Disease: A Systematic Review. Am J Epidemiol 2023; 192 : 305–22. [CrossRef] [Google Scholar]
- Zuccato E, Chiabrando C, Castiglioni S, et al. Estimating community drug abuse by wastewater analysis. Environ Health Perspect 2008; 116 : 1027–32. [CrossRef] [PubMed] [Google Scholar]
- Paul JR, Trask JD, Culotta CS. Poliomyelitic virus in sewage. Science 1939; 90 : 258–9. [CrossRef] [PubMed] [Google Scholar]
- Melnick JL. Poliomyelitis virus in urban sewage in epidemic and in nonepidemic times. Am J Hyg 1947; 45 : 240–53. [Google Scholar]
- Elkana Y, Manaa A, Marzouk Y, et al. Detection of hepatitis A virus in sewage. J Virol Methods 1983; 7 : 259–62. [CrossRef] [PubMed] [Google Scholar]
- Steinmann J. Detection of rotavirus in sewage. Appl Environ Microbiol 1981; 41 : 1043–5. [CrossRef] [Google Scholar]
- Ahmed W, Bertsch PM, Bivins A, et al. Comparison of virus concentration methods for the RT-qPCR-based recovery of murine hepatitis virus, a surrogate for SARS-CoV-2 from untreated wastewater. Sci Total Environ 2020; 739 : 139960. [CrossRef] [PubMed] [Google Scholar]
- Courjon J, Contenti J, Demonchy E, et al. COVID-19 patients age, comorbidity profiles and clinical presentation related to the SARS-CoV-2 UK-variant spread in the Southeast of France. Sci Rep 2021; 11 : 18456. [CrossRef] [PubMed] [Google Scholar]
- Pettit SD, Jerome KR, Rouquie D, et al. ‘All In’: a pragmatic framework for COVID-19 testing and action on a global scale. EMBO Mol Med 2020; 12 : e12634. [CrossRef] [Google Scholar]
- Smyth DS, Trujillo M, Gregory DA, et al. Tracking cryptic SARS-CoV-2 lineages detected in NYC wastewater. Nat Commun 2022; 13 : 635. [CrossRef] [PubMed] [Google Scholar]
- Tierney BT, Foox J, Ryon KA, et al. Towards geospatially-resolved public-health surveillance via wastewater sequencing. Nat Commun 2024; 15 : 8386. [CrossRef] [PubMed] [Google Scholar]
- Child HT, Airey G, Maloney DM, et al. Comparison of metagenomic and targeted methods for sequencing human pathogenic viruses from wastewater. mBio 2023; 14 : e0146823. [CrossRef] [PubMed] [Google Scholar]
- Mac Mahon J, Criado Monleon AJ, Gill LW, et al. Wastewater-based epidemiology (WBE) for SARS-CoV-2 — A review focussing on the significance of the sewer network using a Dublin city catchment case study. Water Sci Technol 2022; 86 : 1402–25. [CrossRef] [PubMed] [Google Scholar]
- Ahmed W, Bivins A, Bertsch PM, et al. Intraday variability of indicator and pathogenic viruses in 1-h and 24-h composite wastewater samples: Implications for wastewater-based epidemiology. Environ Res 2021; 193 : 110531. [CrossRef] [PubMed] [Google Scholar]
- Corpuz MVA, Buonerba A, Vigliotta G, et al. Viruses in wastewater: occurrence, abundance and detection methods. Sci Total Environ 2020; 745 : 140910. [CrossRef] [PubMed] [Google Scholar]
- Kitajima M, Ahmed W, Bibby K, et al. SARS-CoV-2 in wastewater: State of the knowledge and research needs. Sci Total Environ 2020; 739 : 139076. [CrossRef] [PubMed] [Google Scholar]
- Pina S, Jofre J, Emerson SU, et al. Characterization of a strain of infectious hepatitis E virus isolated from sewage in an area where hepatitis E is not endemic. Appl Environ Microbiol 1998; 64 : 4485–8. [CrossRef] [Google Scholar]
- Ye Y, Ellenberg RM, Graham KE, Wigginton KR. Survivability, Partitioning, and Recovery of Enveloped Viruses in Untreated Municipal Wastewater. Environ Sci Technol 2016; 50 : 5077–85. [CrossRef] [PubMed] [Google Scholar]
- Rios G, Lacoux C, Leclercq V, et al. Monitoring SARS-CoV-2 variants alterations in Nice neighborhoods by wastewater nanopore sequencing. Lancet Reg Health Eur 2021; 10 : 100202. [CrossRef] [PubMed] [Google Scholar]
- Treagus S, Lowther J, Longdon B, et al. Metabarcoding of Hepatitis E Virus Genotype 3 and Norovirus GII from Wastewater Samples in England Using Nanopore Sequencing. Food Environ Virol 2023; 15 : 292–306. [CrossRef] [PubMed] [Google Scholar]
- Wurtzer S, Marechal V, Mouchel JM, et al. Evaluation of lockdown effect on SARS-CoV-2 dynamics through viral genome quantification in waste water, Greater Paris, France, 5 March to 23 April 2020. Euro Surveill 2020; 25. [Google Scholar]
- Sherchan SP, Solomon T, Idris O, et al. Wastewater surveillance of Mpox virus in Baltimore. Sci Total Environ 2023; 891 : 164414. [CrossRef] [PubMed] [Google Scholar]
- Wang T, Wang C, Myshkevych Y, et al. SARS-CoV-2 wastewater-based epidemiology in an enclosed compound: A 2.5-year survey to identify factors contributing to local community dissemination. Sci Total Environ 2023; 875 : 162466. [CrossRef] [PubMed] [Google Scholar]
- Cashdollar JL, Wymer L. Methods for primary concentration of viruses from water samples: a review and meta-analysis of recent studies. J Appl Microbiol 2013; 115 : 1–11. [CrossRef] [PubMed] [Google Scholar]
- Michen B, Graule T. Isoelectric points of viruses. J Appl Microbiol 2010; 109 : 388–97. [CrossRef] [PubMed] [Google Scholar]
- Mondal S, Feirer N, Brockman M, et al. A direct capture method for purification and detection of viral nucleic acid enables epidemiological surveillance of SARS-CoV-2. Sci Total Environ 2021; 795 : 148834. [CrossRef] [PubMed] [Google Scholar]
- Vigil K, D’Souza N, Bazner J, et al. Long-term monitoring of SARS-CoV-2 variants in wastewater using a coordinated workflow of droplet digital PCR and nanopore sequencing. Water Res 2024; 254 : 121338. [CrossRef] [PubMed] [Google Scholar]
- Rouchka EC, Chariker JH, Saurabh K, et al. The Rapid Assessment of Aggregated Wastewater Samples for Genomic Surveillance of SARS-CoV-2 on a City-Wide Scale. Pathogens 2021; 10. [PubMed] [Google Scholar]
- Perez-Cataluna A, Cuevas-Ferrando E, Randazzo W, et al. Comparing analytical methods to detect SARS-CoV-2 in wastewater. Sci Total Environ 2021; 758 : 143870. [CrossRef] [PubMed] [Google Scholar]
- Atha DH, Ingham KC. Mechanism of precipitation of proteins by polyethylene glycols. Analysis in terms of excluded volume. Journal of Biological Chemistry 1981; 256 : 12108–17. [CrossRef] [Google Scholar]
- Toribio-Avedillo D, Gomez-Gomez C, Sala-Comorera L, et al. Monitoring influenza and respiratory syncytial virus in wastewater. Beyond COVID-19. Sci Total Environ 2023; 892 : 164495. [CrossRef] [PubMed] [Google Scholar]
- Boogaerts T, Van Wichelen N, Quireyns M, et al. Current state and future perspectives on de facto population markers for normalization in wastewater-based epidemiology: A systematic literature review. Sci Total Environ 2024; 935 : 173223. [CrossRef] [PubMed] [Google Scholar]
- Hsu SY, Bayati M, Li C, et al. Biomarkers selection for population normalization in SARS-CoV-2 wastewater-based epidemiology. Water Res 2022; 223 : 118985. [CrossRef] [PubMed] [Google Scholar]
- Been F, Rossi L, Ort C, et al. Population normalization with ammonium in wastewater-based epidemiology: application to illicit drug monitoring. Environ Sci Technol 2014; 48 : 8162–9. [CrossRef] [PubMed] [Google Scholar]
- Qiu Y, Yu J, Pabbaraju K, et al. Validating and optimizing the method for molecular detection and quantification of SARS-CoV-2 in wastewater. Sci Total Environ 2022; 812 : 151434. [CrossRef] [PubMed] [Google Scholar]
- Wurtz N, Revol O, Jardot P, et al. Monitoring the Circulation of SARS-CoV-2 Variants by Genomic Analysis of Wastewater in Marseille, South-East France. Pathogens 2021; 10. [PubMed] [Google Scholar]
- Garcia-Pedemonte D, Carcereny A, Gregori J, et al. Comparison of Nanopore and Synthesis-Based Next-Generation Sequencing Platforms for SARS-CoV-2 Variant Monitoring in Wastewater. Int J Mol Sci 2023; 24. [PubMed] [Google Scholar]
- Karthikeyan S, Levy JI, De Hoff P, et al. Wastewater sequencing reveals early cryptic SARS-CoV-2 variant transmission. Nature 2022; 609 : 101–8. [CrossRef] [PubMed] [Google Scholar]
- Wilm A, Aw PP, Bertrand D, et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 2012; 40 : 11189–201. [CrossRef] [PubMed] [Google Scholar]
- Grubaugh ND, Gangavarapu K, Quick J, et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol 2019; 20 : 8. [CrossRef] [PubMed] [Google Scholar]
- Caen O, Nizard P, Garrigou S, et al. PCR digitale en micro-compartiments - II. Apport pour la détection quantitative d’ADN tumoral circulant. Med Sci (Paris) 2015 ; 31 : 180–6. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Tisza M, Javornik Cregeen S, Avadhanula V, et al. Wastewater sequencing reveals community and variant dynamics of the collective human virome. Nat Commun 2023; 14 : 6878. [CrossRef] [PubMed] [Google Scholar]
- Smith MF, Maqsood R, Sullins RA, et al. Seasonality of respiratory, enteric, and urinary viruses revealed by wastewater genomic surveillance. mSphere 2024; 9 : e0010524. [CrossRef] [PubMed] [Google Scholar]
- Michael-Kordatou I, Karaolia P, Fatta-Kassinos D. Sewage analysis as a tool for the COVID-19 pandemic response and management: the urgent need for optimised protocols for SARS-CoV-2 detection and quantification. J Environ Chem Eng 2020; 8 : 104306. [CrossRef] [PubMed] [Google Scholar]
- Berchenko Y, Manor Y, Freedman LS, et al. Estimation of polio infection prevalence from environmental surveillance data. Sci Transl Med 2017; 9. [Google Scholar]
- Kumar M, Patel AK, Shah AV, et al. First proof of the capability of wastewater surveillance for COVID-19 in India through detection of genetic material of SARS-CoV-2. Sci Total Environ 2020; 746 : 141326. [CrossRef] [PubMed] [Google Scholar]
- Hellmér M, Paxéus N, Magnius L, et al. Detection of pathogenic viruses in sewage provided early warnings of hepatitis A virus and norovirus outbreaks. Appl Environ Microbiol 2014; 80 : 6771–81. [CrossRef] [Google Scholar]
- Li J, Hosegood I, Powell D, et al. A global aircraft-based wastewater genomic surveillance network for early warning of future pandemics. Lancet Glob Health 2023; 11 : e791–e5. [CrossRef] [PubMed] [Google Scholar]
- Le Targa L, Wurtz N, Lacoste A, et al. SARS-CoV-2 testing of aircraft wastewater shows that mandatory tests and vaccination pass before boarding did not prevent massive importation of omicron variant into Europe. Viruses 2022; 14. [PubMed] [Google Scholar]
Liste des tableaux
Sélection d’études utilisant le séquençage d’acides nucléiques viraux sur des échantillons d’eaux usées. Entérovirus D68 (EVD-68), norovirus groupe II (NoVGII), adénovirus humain 41 (HAdV41), Hépatite A (HAV), virus de la rougeole (MeV), virus de la grippe A (IAV).
Liste des figures
|
Figure 1 Schéma bilan illustrant les étapes du processus de surveillance de la circulation des séquences virales basé sur le séquençage des eaux usées. |
| Dans le texte | |
|
Figure 2 Détails des étapes du processus de surveillance de la circulation des séquences virales basé sur le séquençage des eaux usées. |
| Dans le texte | |
|
Figure 3 Première identification du variant BA.2.86 (Pirola) par analyse des eaux usées de Nice en septembre 2023, effectué à partir d’un prélèvement de la station centrale d’épuration de la Métropole Nice Côte d’Azur, qui collecte les eaux usées d’environ 400 000 habitants (non publié, source IPMC). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.