")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 11, Novembre 2024
|
|
|---|---|---|
| Page(s) | 848 - 857 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2024151 | |
| Published online | 10 décembre 2024 | |
Les nouvelles générations de cellules CAR-T
The new generations of CAR-T cells
1
Dynamics of intracellular organization laboratory, Institut Curie, PSL Research University, Sorbonne Université, CNRS, UMR144, Paris, France
2
Cell therapy Acceleration and Innovation (CellAction), Institut Curie, Suresnes, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
L’approche CAR-T, qui consiste à utiliser des lymphocytes T exprimant un récepteur de l’antigène recombinant (CAR, pour chimeric antigen receptor) permettant d’éliminer les cellules présentant un antigène particulier, est l’une des immunothérapies les plus prometteuses pour soigner les cancers. L’ingénierie des CAR-T a évolué au fil de leurs générations, pour renforcer leur activité et leur spécificité, et surmonter leurs limites, comme leur faible persistance, leur toxicité et leur inefficacité dans la lutte contre les tumeurs solides. Cette revue explore les différentes générations de CAR, les tests cliniques en cours sur le cancer et les maladies auto-immunes, ainsi que les limites associées aux cellules CAR-T dans le traitement des cancers.
Abstract
Chimeric antigen receptor (CAR)-T is one of the most promising modern cancer immunotherapies. In the recent years, impressive results have been obtained in the treatment of cancer which led to FDA approval for the treatment of liquid tumors. In this cell-based therapy, immune cells (e.g. T and NK cells) are engineered to express a synthetic receptor CAR to specifically recognize and eliminate cells expressing a target antigen.
CAR has evolved through different generations aiming to boost its biological activity and overcome limitations such as low persistence, limited potency, life-threatening toxicity and inefficient activity against solid tumor. The present review provides an overview of the different CAR generations, starting from the 1st generation with limited cytotoxic activity until the latest generation, the 5th generation or new generation, developed to overcome various limitations of CAR T therapy. The current ongoing clinical trials in cancer and autoimmune diseases, and the limitation associated with CAR-T cells in cancer therapy, are also discussed.
© 2024 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (http://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (http://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Zélia Gouveia).
Le cancer est un problème majeur de santé publique dans le monde, et est parmi les deux premières causes de décès dans nombreux pays. Selon l’Organisation mondiale de la santé (OMS), plus de dix-neuf millions de nouveaux cas ont été enregistrés en 2020, et près de dix millions de personnes sont décédées à la suite d’un cancer. Les recherches réalisées pour lutter contre cette maladie ont donné naissance à différentes thérapies anticancéreuses qui viennent compléter ou éviter la chirurgie, comme les chimiothérapies, les radiothérapies et les biothérapies. Actuellement, les biothérapies anticancéreuses englobent des approches multiples, comme les immunothérapies (anticorps anti-point de contrôle immunitaire, cellules CAR-T), les thérapies géniques (adénovirus recombinant) [1] et les vaccins anticancéreux (vaccins conventionnels, à ARN, ou à base de cellules dendritiques autologues) [2]. Les immunothérapies cellulaires, en particulier celles fondées sur l’utilisation de lymphocytes T (LT) exprimant un récepteur antigénique chimérique (CAR), nommées cellules CAR-T, révolutionnent le traitement de certains cancers, en particulier les lymphomes et certaines leucémies aigües [3].
Le complexe majeur d’histocompatibilité de classe I (CMH I), constitué d’un groupe de molécules membranaires exprimées par les cellules1, est indispensable au processus de présentation des antigènes aux lymphocytes, une étape clé dans la réponse immunitaire exercée par les LT cytotoxiques, les lymphocytes participant à la lutte contre les cellules cancéreuses. L’inhibition ou la perte d’expression des molécules du CMH I dans les cellules cancéreuses est considérée comme l’un des moyens utilisés par les cellules tumorales pour échapper aux LT [4]. Le CAR est un récepteur recombinant conçu pour reconnaître un antigène cible spécifique indépendamment du CMH I. Il permet de rediriger et d’activer les cellules immunitaires afin de reconnaître et d’éliminer les cellules exprimant cet antigène particulier (Figure 1). Dans cette revue, nous décrirons la structure des CAR et leur évolution au travers des différentes générations développées pour améliorer l’efficacité, la persistance et de la sécurité de ces cellules. Nous préciserons également le mode d’action des CAR et présenterons les dernières applications cliniques et les limites rencontrées pour assurer un plus large accès à cette thérapie eu égard son coût, et son application dans le traitement contre les cancers solides.
 |
Figure 1. Structure des CAR. Les CAR sont généralement composés de trois domaines : un domaine extracellulaire permettant de reconnaître un antigène à la surface des cellules tumorales ; un domaine transmembranaire pour assurer sa stabilité et son expression à la surface des lymphocytes T ; et un domaine intracellulaire de signalisation permettant d’émettre des signaux de co-stimulation et d’activer les lymphocytes T. |
Évolution et structure des CAR
L’immunothérapie cellulaire adoptive a été introduite pour la première fois dans les années 1980, dans une approche où les lymphocytes infiltrant la tumeur étaient collectés et amplifiés afin d’être utilisés pour soigner les patients présentant un cancer avancé [5, 6]. Au début des années 1990, des outils génétiques ont été mis au point pour élaborer des cellules immunitaires, comme les LT [7], exprimant des molécules étrangères, et en 1989, l’immunologiste Zelig Eshhar réalise, pour la première fois, un récepteur recombinant capable de reconnaître un antigène tumoral et d’activer les LT [8, 9]. Il s’agissait là de la première génération de CAR, dont l’activité in vivo restait cependant limitée.
Alors que, de façon naturelle, la reconnaissance du peptide antigénique repose sur la présentation du complexe peptide-CMH au récepteur de l’antigène des LT (TCR, T cell receptor), dans le contexte d’un CAR, les cellules immunitaires reconnaissent directement et spécifiquement l’antigène présent à la surface des cellules tumorales, indépendamment de la présentation du CMH, grâce à sa liaison au fragment d’anticorps qui compose la partie extracellulaire du CAR (par exemple un fragment scFv [single-chain variable fragment] ou un anticorps simple chaîne/Nanobody®). Le scFv est constitué du domaine variable de la chaîne lourde (VH) et du domaine variable de la chaîne légère (VL) d’un anticorps ayant une spécificité anti-tumorale, reliés par une séquence d’acides aminés hydrophile, flexible (la plus utilisée étant la séquence (Gly4Ser)x3). Le scFv est associé à un domaine transmembranaire par une charnière/espaceur (dérivée par exemple de CD8 ou de la région charnière [hinge] des IgG1/4) qui lui apporte de la flexibilité, augmentant ainsi sa capacité de reconnaissance des antigènes. Le domaine transmembranaire est une hélice alpha hydrophobe, généralement dérivée de celle de protéines transmembranaires, telles que CD8, CD28 ou CD4, qui assure la stabilité et l’expression du CAR à la surface des cellules. La région intracellulaire du CAR est composée de domaines de costimulation associés à des domaines d’activation, nécessaires à la survie, la persistance, la cytotoxicité et à d’autres activités des cellules immunitaires. Les domaines de costimulation sont généralement dérivés de l’un des domaines intracellulaires de costimulation des protéines 4.1BB, CD28, ICOS, CD27, MYD88, CD40 ou OX40, tandis que le domaine d’activation correspond à la région de CD3ζ qui contient trois motifs ITAM (immunoreceptor tyrosine-based activation motif) nécessaires à l’initiation de la cascade d’activation cytotoxique cellulaire (Figure 1).
De nouvelles générations CAR-T ont ensuite été développées, se montrant d’une efficacité de plus en plus importante dans l’élimination des tumeurs. Ceci a conduit, en 2017, à l’autorisation par la FDA (Food and Drug Administration) de la première thérapie CAR-T ciblant la protéine CD19 présente à la surface des lymphocytes B, pour traiter la leucémie lymphoblastique aigüe pédiatrique récurrente et réfractaire [10]. Aujourd’hui, six thérapies CAR-T ont été approuvées pour le traitement des tumeurs dites « liquides ». L’application des thérapies CAR-T dans les tumeurs solides reste néanmoins un défi et de nouvelles générations de CAR-T sont régulièrement développées pour améliorer ou surmonter les limites rencontrées (Figure 2).
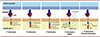 |
Figure 2. Les générations de CAR. La reconnaissance de la cible tumorale est en général assurée par un scFv (bien que le même rôle puisse être joué par d’autres domaines de liaison). Le rectangle rouge représente le domaine d’activation (dérivé de la protéine CD3zeta [CD3ζ]) ; l’ovale bleu et le losange vert représentent les domaines de co-stimulation. L’étoile rose représente l’inducteur moléculaire, la flèche et le carré bleu clair symbolisent l’expression d’un gène, et les points bleu clair des cytokines qui peuvent être sécrétées. Le triangle jaune est le domaine IL-2Rβ nécessaire à l’activation de la voie JAK-STAT3/5. |
Les générations de CAR
Première génération
La première génération de CAR, appelée T-bodies, a été développée par l’équipe de Z. Eshhar [8, 9]. Elle était composée d’un domaine extracellulaire pour la reconnaissance de l’antigène tumoral, et d’un domaine transmembranaire et d’un domaine intracellulaire pour l’activation des LT (Figure 2). Cette première génération de cellules CAR-T s’est avérée efficace pour éliminer les tumeurs in vitro, mais dans les essais cliniques, elle s’est révélée avoir une prolifération et une persistance limitées, ce qui limitait son efficacité pour le contrôle des tumeurs [11, 12]. Une cause probable expliquant cette activité limitée est que les LT naïfs ont besoin d’une double signalisation pour être activés en cellules effectrices. Ces signaux comprennent en effet le signal donné par la reconnaissance par le TCR (CD3ζ) du LT du complexe antigénique peptide-MHC (p-MHC) exprimé par la cellule cible, mais aussi un signal d’activation donné par les cellules présentatrices d’antigènes au LT via des molécules comme CD80 (B7.1) et CD86 (B7.2).
Seconde génération
La deuxième génération de CAR visait à améliorer la persistance et la prolifération limitées de la première génération, en introduisant un domaine de co-stimulation dans la région intracellulaire du CAR (Figure 2). L’ajout de ce domaine de co-stimulation, dérivé d’OX-40, de 4-1BB ou de CD27, a permis d’améliorer la prolifération, la survie, la cytotoxicité et la réponse soutenue des CAR-T in vivo. Les essais cliniques utilisant cette deuxième génération de CAR-T ont montré des résultats impressionnants dans le traitement des hémopathies malignes (lymphome, leucémie et myélome), ce qui a conduit à l’approbation de certains d’entre eux par la FDA, pour le traitement de ces tumeurs. Alors que ces CAR-T ont montré une très bonne efficacité pour le traitement des tumeurs liquides, leur efficacité reste encore très limitée dans les tumeurs solides, principalement en raison d’une infiltration limitée des LT dans la tumeur, et d’un microenvironnement tumoral hostile à l’entrée des CAR-T [13].
Troisième génération
Une troisième génération de CAR-T a été imaginée pour surmonter les limitations rencontrées. Cette génération de CAR repose sur l’ajout d’un domaine intracellulaire costimulateur supplémentaire dans le but d’améliorer l’efficacité des cellules CAR-T. Les principales combinaisons utilisées sont ainsi CD28/4-1BB/CD3ζ ou CD28/OX40/CD3ζ. L’objectif était de tirer parti des caractéristiques spécifiques de ces différents domaines, par exemple, en combinant CD28, qui favorise l’expansion rapide des LT et une toxicité plus élevée, avec 4-1BB, connu pour être associé à une persistance plus longue [14]. Dans l’un des premiers essais cliniques, cette génération de CAR-T n’a pas apporté d’avantages significatifs dans le traitement du lymphome [15]. Cependant, d’autres études, y compris des essais cliniques visant à mieux comprendre si cette génération était plus efficace que la seconde génération, ont rapporté une meilleure expansion et une meilleure persistance des CAR-T de troisième génération [16]. Dans les hémopathies malignes, ces CAR-T ont confirmé leur moindre toxicité associée à une efficacité clinique [17], même si d’autres essais cliniques n’ont pas mis en évidence de différences significatives entre les deux générations [18]. L’avantage de la troisième génération de CAR-T reste à confirmer et d’autres études sont encore nécessaires pour démontrer sa supériorité et pour mieux comprendre quelle serait la combinaison de signaux « idéale » pour promouvoir l’activité et la survie des LT tout en évitant leur épuisement, leur prolifération limitée et leur mort prématurée.
La quatrième génération : les TRUCK
Même si certains auteurs considèrent qu’elles forment ensemble la dernière génération de CAR [19], nous avons choisi de séparer la quatrième génération de CAR, ou « TRUCK » (T cells redirected for universal cytokine killing) de la 5e génération.
Cette quatrième génération de cellules CAR-T combine les CAR de deuxième génération et la capacité de la cellule à libérer, de façon constitutive ou après induction, un facteur trophique, tel qu’une cytokine. L’action additionnelle de ces facteurs permet d’augmenter l’efficacité, la survie et la persistance des cellules CAR-T, et de modifier le microenvironnement tumoral pour permettre un meilleur accès à la tumeur ou contrer l’immunosuppression [20]. Les facteurs trophiques les plus couramment utilisés sont des cytokines pro-inflammatoires, notamment l’IL-7, l’IL-12, l’IL-18, l’IL-21 et l’lL-23, qui stimulent le système immunitaire en créant un environnement plus fortement antitumoral et en améliorant la persistance, la survie et la cytotoxicité des cellules CAR-T [21]. Plusieurs essais cliniques visent à évaluer cette génération de CAR-T, notamment les cellules CAR-T co-exprimant l’IL-18 (NCT04684563), l’IL-12 (NCT03542799) ou l’IL-7, en combinaison avec la chimiokine CCL19 (chemokine (C-C motif) ligand 19) (NCT04381741, NCT03929107, NCT04833504, NCT03778346). Dans cette dernière combinaison, alors que l’IL-7 favorise la prolifération et la survie des LT, la chimiokine CCL19 agit comme un chemo-attractant des LT et des cellules dendritiques. Ces essais seront essentiels pour évaluer, et éventuellement confirmer, la supériorité de ce type de CAR.
La cinquième génération ou nouvelle génération
Malgré les succès cliniques observés avec les dernières générations de CAR, persistent encore plusieurs limitations et inconvénients, restreignant leur application thérapeutique, principalement leur efficacité limitée dans les tumeurs solides et la toxicité qui leur est associée. Les nouvelles générations tentent de répondre à ces limitations en apportant des innovations fondées sur l’édition du génome et la biologie synthétique. L’un des exemples de cinquième génération repose, une fois de plus, sur le couplage d’un CAR de seconde génération avec un domaine cytoplasmique tronqué de la chaîne β du récepteur de l’IL-2 contenant le site de liaison du facteur de transcription STAT3. Il en résulte une activation simultanée de CD3ζ, le domaine de costimulateur, et la voie de signalisation JAK-STAT3/5, qui accroît la cytotoxicité et la persistance des LT [22].
Plusieurs améliorations sont envisagées pour aboutir à des cellules CAR-T plus spécifiques, plus puissantes, modulables et plus sûres. Il s’agit notamment des systèmes « dual/multi-targeting », « splitable », « inducible (SynNotch CAR) », « Small molecule protease-based regulation (SWIFF) CAR », « inhibitory CAR », et « suicide-switch CAR » (Figure 3) [23, 24]. Les CAR universels sont un autre système qui suscite un intérêt croissant (Figure 3) [25], mais l’applicabilité thérapeutique de ces CAR reste à étudier.
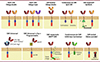 |
Figure 3. Quelques exemples des nouvelles générations de CAR. Parmi les CAR de nouvelle génération, certains sont capables de reconnaître deux ou plusieurs antigènes tumoraux distincts, soit par l’expression simultanée de plusieurs CAR, soit par l’utilisation de séquences de liaison ciblant plusieurs antigènes (porte logique « OR »). Les « split CAR » consistent à séparer sur deux CAR, le domaine d’activation et le domaine de signalisation, pour la reconnaissance de deux antigènes nécessaires à l’activation optimale des cellules CAR -T (porte logique « AND »). Dans les « SynNotch CAR », il est nécessaire que le récepteur Notch soit engagé lors de la reconnaissance de l’antigène (signal scFv 1) pour la transcription intracellulaire d’un CAR fonctionnel. Les cellules « Universal CAR » ont pour but d’assurer la polyvalence de la reconnaissance des antigènes tumoraux par un système intermédiaire. Ce système intermédiaire consiste en la production d’un CAR universel avec un scFv qui reconnaîtra une étiquette fusionnée à un scFv spécifique de différents antigènes tumoraux. Dans le cas du « split Universal Programmable CAR », le CAR est séparé en zipCAR (domaine de costimulation et d’activation) et zipFv (scFv pour la reconnaissance de l’antigène) liés par complémentarité pour former un CAR fonctionnel. Le système de « Small molecule protease-based regulation (SWIFF) CAR » est composé d’un CAR fusionné à son extrémité C-terminale à un site cible de protéase, d’une protéase et d’un composant de dégradation des protéines (degron). La protéase est active dans des conditions normales avec la production d’un CAR fonctionnel. Lors de l’administration d’un inhibiteur de la protéase (par exemple l’asunaprevir), le degron est conservé, ce qui entraîne la dégradation du CAR. Dans le système « inhibitor CAR », le CAR est exprimé simultanément avec un autre récepteur contenant un signal inhibiteur qui reconnaît les tissus sains et empêche l’activité de la cellule CAR-T. Un autre système de sécurité dans le CAR est l’incorporation de gènes ou de commutateurs de suicide contrôlables (par exemple, iCasp9), coexprimés avec le CAR pour tuer les cellules CAR-T en cas d’effets secondaires graves. |
La cellule CAR-T allogénique est une option prometteuse visant à surmonter certains des principaux défis que posent les CAR-T autologues : notamment, la standardisation du produit CAR-T, son coût de production et la disponibilité immédiate des cellules CAR-T cryoconservées pour plusieurs patients, ainsi que la réduction ou l’absence de risque d’échec de la fabrication. Les thérapies allogéniques présentent néanmoins un risque élevé de maladie du greffon contre l’hôte (GVHD, pour graft versus host disease), une réponse qui survient lorsque des cellules immunitaires allogéniques sont greffées et reconnaissent les cellules de l’hôte comme étrangères et les détruisent. Inversement, les cellules CAR-T allogéniques peuvent être la cible du système immunitaire de l’hôte et être détruites. Plusieurs stratégies sont actuellement à l’étude pour prévenir ces risques [26]. Une alternative pour prévenir ou minimiser la réaction de GVHD est d’éliminer dans les LT greffés, le TCR (récepteur endogène des lymphocytes T, constitué des chaînes αβ), par édition du génome des cellules en appliquant la technique CRISPR/Cas9. Une autre option intéressante consiste en l’intégration des gènes codant le CAR dans le locus TRAC (TCR alpha constant) codant la chaîne α du TCR, ce qui permet une intégration non aléatoire, une expression uniforme du CAR (promoteur endogène), une signalisation tonique plus faible, une amélioration de l’activité des lymphocytes T cytotoxiques, tout en inactivant le TCR du greffon, comme l’a montré l’étude d’Eyquem et al. utilisant des cellules CD19-CAR-T [27].
Mode d’action des cellules CAR-T (Figure 4)
 |
Figure 4. Mode d’action d’une cellule CAR-T. La partie extracellulaire du CAR se lie spécifiquement à l’antigène exprimé à la surface des cellules tumorales, ce qui active le recrutement de la protéine ZAP70 par le domaine d’activation (CD3ζ) et le déclenchement d’une cascade d’activation. Le domaine de co-stimulation permet d’activer d’autres cascades de signalisation (PI3K dans le cas de CD28, et TRAF1/2 dans le cas de 4-1BB). Ces cascades agissent sur les voies contrôlées par NF-κB et STAT3, induisant par exemple la production de cytokines telles que l’IL-2, l’IL-5 et l’IFNγ. L’ensemble de ces réactions favorisent l’expansion et la toxicité des lymphocytes T, notamment par la libération de perforine et de granzyme. L’introduction de granzyme dans la cellule tumorale, via les pores formés par la perforine, induit la mort de la cellule. |
La reconnaissance de la tumeur par les cellules CAR-T induit une cascade de signaux qui conduisent à la prolifération, à la persistance et à la cytotoxicité de ces cellules. Il existe une variété de domaines de co-stimulation intégrés dans les différents CAR qui ont un effet déterminant sur le phénotype et la performance des LT. Les CAR, dans leur majorité, sont composés de domaines de co-stimulation dérivés de CD28 ou de 4-1BB.
CD28 appartient aux récepteurs de co-stimulation des LT. C’est par son motif YMNM (Tyr-Met-Asp-Met) après la phosphorylation de la tyrosine, que se fixent et s’activent la sous-unité p85 de la phosphoinositide 3-kinase (PI3K) et la protéine adaptatrice Grb2, induisant la prolifération du LT et la production d’IL-2. 4-1BB est un co-stimulateur qui favorise le recrutement des protéines associées aux récepteurs TRAF (TNF receptor associated factor) 1, 2 et 3 dans son domaine intracellulaire. Cela mène finalement à l’activation de la voie de signalisation NF-NB (nuclear factor-kappa B) qui conduit, notamment, à la production d’IL-2 et favorise la survie et la prolifération des LT.
L’engagement du CAR induit la phosphorylation des trois domaines ITAM du CD3] par la famille de tyrosine kinases Src, favorisant le recrutement de la tyrosine kinase ZAP-70 (zeta-chain-associated protein of 70 kDa) / Syk (spleen tyrosine kinase). ZAP-70 est ensuite stimulée par Lck (lymphocyte cell-specific protein-tyrosine kinase), favorisant la signalisation en aval qui active les facteurs de transcription NF-NB, NFAT (nuclear factor of activated T-cells) et AP-1 (activating protein-1), nécessaires à la transcription des gènes codant les protéines impliquées dans l’activation, l’inflammation et la cytotoxicité des LT [28].
Les applications cliniques des cellules CAR-T
Les cellules CAR-T sont principalement destinées au traitement des cancers mais, ces dernières années, d’autres applications ont été envisagées pour le traitement, par exemple, de maladies autoimmunes [22]. Plusieurs essais cliniques utilisant des cellules CAR-T dirigées contre différents antigènes associés aux tumeurs mais également exprimés par des cellules non tumorales évaluent aussi l’intérêt de leur utilisation pour traiter certaines maladies auto-immunes (Tableau I).
Exemples d’essais cliniques portant sur les cellules CAR-T (Clinicaltrials.gov). BCMA : antigène de maturation des cellules B (B-cell maturation antigen) ; CEA : antigène carcinoembryonnaire (carcinoembryonic antigen) ; GFRα4 : récepteur alpha-4 de la famille GDNF (GDNF family receptor Dα), ICAM/CD54 : molécule d’adhérence intercellulaire 1 (intercellular adhesion molecule 1). * : République populaire de Chine.
Cibler les cellules tumorales
En 2006, des chercheurs néerlandais ont publié pour la première fois un article relatant un essai clinique évaluant des cellules CAR-T dirigées contre le cancer rénal métastatique, essai où avait été observée une toxicité hépatique sans réponse efficace contre la maladie [29]. Un résultat décevant mais une étude détaillée ultérieure démontra que cette toxicité était liée à une activité des cellules CAR-T dépendant de l’antigène tumoral ciblé, suggérant ainsi que l’utilisation de ces cellules CAR-T pouvait présenter un intérêt, si cette toxicité était contrôlée [30].
Après plus de 10 ans, en 2017, le premier traitement par cellules CAR-T-CD19 fut approuvé par la FDA, indiqué pour le traitement de certaines formes de tumeurs malignes à cellules B. À ce jour, il existe six traitements fondés sur des cellules CAR-T de deuxième génération contre des hémopathies malignes, telles que la leucémie lymphoblastique aiguë à cellules B (LLA-B) et le myélome multiple (MM).
En ce qui concerne les tumeurs solides, aucun essai clinique n’a atteint la phase III, illustrant la difficulté qu’il y a à cibler efficacement les tumeurs solides, et le besoin de mieux comprendre les mécanismes de résistance et d’échappement de ces tumeurs vis-à-vis des cellules CAR-T [13, 31]. Plusieurs candidats ont donné néanmoins récemment des résultats prometteurs pour traiter certaines tumeurs solides [31]. Selon le site Clinicaltrials.gov, plus de dix essais cliniques portent sur l’efficacité et la sécurité de cellules CAR-T anti-HER2 (human epidermal growth factor receptor-2) pour traiter des patients présentant un cancer dont les cellules tumorales expriment HER2 (par exemple, le cancer du sein, le cancer gastrique et les glioblastomes). D’autres cibles en cours d’évaluation pour le traitement de tumeurs solides comprennent le récepteur du facteur de croissance épidermique (EGFR), la variante III de l’EGFR (EGFRvIII) et la chaîne α2 du récepteur de l’IL-13 (IL-13Rα2), surexprimés dans les glioblastomes. La mésothéline, qui est exprimée dans plusieurs tumeurs solides (le mésothéliome, le cancer épithélial de l’ovaire, l’adénocarcinome canalaire pancréatique, les cancers du poumon, de l’utérus, du sein triple négatif, gastrique), est également une cible prometteuse [31].
Les autres applications cliniques
Les applications cliniques des CAR-T ne se limitent pas seulement aux cancers. Des résultats in vitro ont permis de proposer les CAR-T comme une piste thérapeutique intéressante pour traiter certaines maladies auto-immunes [32]. Un essai clinique incluant des patients présentant des maladies auto-immunes, comme le lupus érythémateux disséminé, la sclérodermie systémique, ou la myosite inflammatoire idiopathique, a donné des résultats très prometteurs. Dans cette étude, tous les patients ont en effet répondu favorablement au traitement avec toutefois quelques effets secondaires, principalement un syndrome de relargage de cytokines (SRC) de grade 1. Néanmoins, chez l’un des patients, un SRC de grade 2 accompagné d’un syndrome de neurotoxicité associé aux cellules effectrices immunitaires (ICANS, pour immune effector cell-associated neurotoxicity syndrome) de grade 1 ont été observés. Une pneumonie a nécessité l’hospitalisation de ce patient [33]. Cet essai a permis cependant de démontrer l’efficacité des cellules CAR-T pour le traitement des maladies autoimmunes avec un degré de sécurité acceptable. Il existe actuellement plusieurs essais cliniques visant à démontrer l’efficacité et la sécurité de ce type de traitement.
Les limites actuelles
Comme nous l’avons vu, l’application thérapeutique des cellules CAR-T peut être associée à des effets indésirables parfois graves, qui peuvent mettre la vie du patient en danger. Nous revenons ici sur les effets secondaires majeurs.
Le syndrome de relargage de cytokines
Le syndrome de relargage de cytokines (SRC) constitue la forme de toxicité la plus fréquente associée aux traitements par cellules CAR-T. Il se manifeste par une réaction inflammatoire généralisée provoquée par une production excessive de cytokines inflammatoires [34]. La réaction commence par une production massive de cytokines, telles que l’IL-6, l’IFN(interféron)-γ, le TNF-α (tumor necrosis factor alpha), le GM-CSF (granulocyte-macrophage colonystimulating factor), l’IL-2, l’IL-8 et l’IL-10, qui est suivie d’une réponse inflammatoire secondaire causée par l’activation des cellules présentatrices d’antigènes (les cellules dendritiques, les monocytes, les macrophages, les lymphocytes B). L’ensemble du processus conduit à une production élevée de cytokines inflammatoires, dont l’IL-6, considérée comme le principal facteur de toxicité du SRC. En effet, l’IL-6 stimule la production de cytokines par plusieurs cellules immunitaires, ce qui entraîne, en cascade, une libération accrue de facteurs inflammatoires. Les symptômes les plus fréquents associés au SRC sont une forte fièvre, de légers symptômes pseudo-grippaux, des problèmes rénaux et des chutes de tension artérielle, potentiellement mortelles dans les cas les plus graves. Il est nécessaire de maintenir une surveillance périodique ou constante de la fonction cardiovasculaire et de la température chez les patients traités par des cellules CAR-T afin d’agir précocement et d’éviter des complications létales. Le tocilizumab, un anticorps antagoniste qui se lie au récepteur de l’IL-6 et l’inhibe, est utilisé pour éviter les formes graves de SRC [35].
Le syndrome de neurotoxicité associé aux cellules effectrices immunitaires
Un autre effet secondaire important induit par les traitements par cellules CAR-T est l’ICANS. Ce syndrome d’encéphalopathie est probablement la conséquence de la libération massive de cytokines. La physiopathologie de l’ICANS n’est toujours pas bien comprise, mais ce syndrome survient en général à la suite d’un SRC à l’origine de l’activation des cellules endothéliales, de la rupture de la barrière hémato-encéphalique et de l’infiltration importante de cellules immunitaires et de cytokines inflammatoires dans le cerveau [36], l’ensemble provoquant de graves lésions du système nerveux central qui sont aggravées par l’infiltration de cellules CAR-T [37]. Les troubles neurologiques liés à ce syndrome les plus communs sont des maux de tête, des vertiges, un délire et une altération des capacités cognitives. L’évaluation de la sévérité de ces troubles se fonde actuellement sur le score défini par l’ASTCT (American Society for Transplantation and Cellular Therapy), un score allant de 1 à 4 selon les effets observés chez le patient. Le traitement peut aller d’un simple soin de support jusqu’aux soins intensifs et à la réanimation dans les cas les plus graves.
L’atteinte d’une cible spécifique hors-tumeur
L’effet « sur cible/hors tumeur » (on-target/off-tumor) est l’effet indésirable d’une thérapie dont la cible est exprimée par les cellules cancéreuses mais aussi par des cellules saines. Le CAR ne distinguant pas les cellules tumorales des tissus sains, les cellules CAR-T qui l’expriment détruisent les cellules présentant l’antigène ciblé, qu’elles soient tumorales ou saines. Cela représente une limite majeure dans l’application cliniques des cellules CAR-T et souligne l’importance d’identifier des antigènes qui soient spécifiques des tumeurs. Cela est particulièrement évident concernant les tumeurs solides, pour lesquelles le nombre d’antigènes spécifiques reste limité, et une élimination de cellules saines, même temporaire, comme dans le cas des cellules CAR-T anti-CD19, ne peut être envisagée. Lors d’un essai clinique, par exemple, le traitement qui reposait sur des cellules CAR-T dirigées contre HER2 a causé chez les patients, une toxicité mortelle, vraisemblablement à cause de la reconnaissance de HER2 exprimé par les cellules pulmonaires normales, conduisant à un SRC, suivi d’une défaillance multi-organes [38].
Échappement par perte de l’antigène
L’efficacité des thérapies par cellules CAR-T dépend de l’expression par les cellules tumorales de l’antigène ciblé. Elle est donc fortement diminuée lorsque l’expression de cet antigène par les cellules cibles se réduit. Cette réduction d’expression a été décrite comme un mécanisme courant de résistance tumorale. En effet, dans de nombreux essais cliniques, une large fraction des cellules qui échappent et résistent au traitement se révèlent avoir subi une perte partielle ou totale d’expression de l’antigène ciblé. Ce phénomène a été observé, par exemple, chez des patients présentant une LAL B en rechute et/ou réfractaire, avec une régulation négative ou une perte de l’antigène CD19 observée chez 40 % des patients [39]. Des données de suivi récentes suggèrent que cette perte d’antigène est un mécanisme commun de résistance aux cellules CAR-T. Ce mécanisme de résistance a également été observé chez des patients présentant un cancer dit « solide ». Des approches alternatives sont en cours de développement pour permettre aux cellules CAR-T de cibler durablement les tumeurs et être moins sensibles à ces mécanismes de résistance, avec, par exemple, l’utilisation de CAR multi-spécifiques (Dual or tandem CARs) (Figure 3) ou la combinaison avec des immunothérapies monospécifiques.
Le microenvironnement tumoral immunosuppressif
Un microenvironnement tumoral (MT) immunosuppressif se caractérise par un environnement complexe, dynamique et réfractaire constitué principalement de cellules immunitaires, de cellules tumorales, de fibroblastes et de cellules endothéliales, ainsi que d’autres structures et molécules (par exemple, la matrice extracellulaire, des protéases, des microvésicules). Le MT est par ailleurs hypoxique, pauvre en nutriments, avec un pH faible et une concentration importante d’espèces réactives de l’oxygène. Le MT favorise ainsi la survie des cellules tumorales tout en entravant l’activité anti-tumorale des cellules immunitaires dont les lymphocytes T. Dans le MT, les cellules suppressives d’origine myéloïde (myeloidderived suppressor cells, ou MDSC), les macrophages associés à la tumeur (tumor-associated macrophage, ou TAM) et les lymphocytes T régulateurs (Treg) qui sont présentes, peuvent également induire une immunosuppression par la production de cytokines et de facteurs de croissance. L’hypoxie du MT favorise aussi l’expression de points de contrôle immunitaire (PD-L1 [programmed death-ligand 1] par les cellules tumorales, ou CTLA-4 [cytotoxic t-lymphocyte-associated protein-4] par les Treg et les TAM), ce qui conduit au blocage de l’activité des LT. La réponse des cellules CAR-T contre les cellules tumorales est ainsi diminuée ou bloquée en raison de leurs faibles expansion et infiltration, et de leur courte persistance dans cet environnement [40]. La combinaison d’inhibiteurs de points de contrôle immunitaires avec des cellules CAR-T peut donc conduire à une réponse anti-tumorale plus importante avec une persistance et une activité améliorées des LT. De nouvelles générations de cellules CAR-T ont été générées afin de surmonter ces limitations [40].
Conclusion
L’approche par CAR a révolutionné le domaine des thérapies cellulaires contre les cancers, avec des résultats impressionnants dans le traitement de certaines tumeurs. Les générations successives de CAR ont permis d’améliorer l’efficacité des traitements tout en réduisant leur toxicité. La réussite des CAR dans le domaine des cancers hématologiques est aujourd’hui considérée comme une avancée majeure dans le domaine des biothérapies. Néanmoins, plusieurs limitations importantes persistent pour l’utilisation et le développement de ce type de thérapies. Il s’agit notamment d’effets secondaires qui peuvent mettre la vie du patient en danger. Leur application pour le traitement des cancers solides reste également limitée, très probablement en raison d’une persistance limitée des cellules CAR-T et de la difficulté d’identifier des cibles spécifiques. De plus, les mécanismes d’échappement, notamment la perte d’expression de l’antigène par ces cellules tumorales, ou l’action immunosuppressive de l’environnement tumoral, affectent l’efficacité des cellules CAR-T.
Pour surmonter ces limitations, d’autres générations de cellules CAR-T ont été développées. Il s’agit notamment de la troisième génération de CAR, intégrant un deuxième domaine de costimulation, suivie de la quatrième génération, ou TRUCK, et, plus récemment, de la mise au point de nouvelles générations apportant plus de contrôle. La recherche reste très active pour améliorer les stratégies CAR, incluant l’utilisation d’autres cellules immunitaires (comme les cellules NK [natural killer], ou les macrophages). De nombreux essais cliniques sont en cours, ciblant des tumeurs hématologiques, des tumeurs solides et des maladie auto-immunes. Le potentiel de ces approches est très important et représente un réel espoir, en particulier pour le traitement des cancers et de l’auto-immunité.
Remerciements
Nous souhaitons remercier l’équipe du laboratoire « Dynamics of the Intra-cellular », UMR144 de l’Institut Curie, pour les discussions scientifiques stimulantes. Nous voulons également remercier l’équipe CellAction à l’Institut Curie, Suresnes. Djamel Messaoudi bénéficie d’une bourse de doctorat financée par le DIM Bionconvergence pour la Santé (DIM BioConvS) de la région Île-de-France. Le développement de CART modulables in vivo est soutenu par le financement ANR-22-CE14-0066-01.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Les molécules du CMH de classe I (ou HLA pour human leucocyte antigen) sont composées de deux chaînes polypeptidiques : une chaîne α longue responsable de la reconnaissance de l’antigène et une chaîne β courte, appelée β2-microglobuline. Ces molécules sont exprimées par toutes les cellules nucléées
Références
- Singh V, Khan N, Jayandharan GR. Vector engineering, strategies and targets in cancer gene therapy. Cancer Gene Ther 2022 ; 29 : 402–17. [CrossRef] [PubMed] [Google Scholar]
- Tanyi JL, Bobisse S, Ophir E, et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med 2018 ; 10 : eaao5931. [CrossRef] [PubMed] [Google Scholar]
- June CH, O’Connor RS, Kawalekar OU, et al. CAR T cell immunotherapy for human cancer. Science 2018 ; 359 : 1361–5. [CrossRef] [PubMed] [Google Scholar]
- Paulson KG, Voillet V, McAfee MS, et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun 2018 ; 9 : 3868. [Google Scholar]
- Rosenberg SA, Eberlein TJ, Grimm EA, et al. Development of long-term cell lines and lymphoid clones reactive against murine and human tumors: a new approach to the adoptive immunotherapy of cancer. Surgery 1982 ; 92 : 328–36. [PubMed] [Google Scholar]
- Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986 ; 233 : 1318–21. [CrossRef] [PubMed] [Google Scholar]
- Sadelain M. Methods for retrovirus-mediated gene transfer into primary T-lymphocytes. Methods Mol. Med. 1997 ; 7 : 241–8. [Google Scholar]
- Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 1989 ; 86 : 10024–8. [CrossRef] [PubMed] [Google Scholar]
- Eshhar Z, Waks T, Gross G, et al. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A 1993 ; 90 : 720–4. [CrossRef] [PubMed] [Google Scholar]
- Sadelain M. CD19 CAR T Cells. Cell 2017 ; 171 : 1471. [CrossRef] [PubMed] [Google Scholar]
- Park JR, Digiusto DL, Slovak M, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther 2007 ; 15 : 825–33. [Google Scholar]
- Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 2006 ; 12 : 6106–15. [CrossRef] [PubMed] [Google Scholar]
- Marofi F, Motavalli R, Safonov VA, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021 ; 12 : 81. [CrossRef] [Google Scholar]
- Stegen SJC van der, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov 2015 ; 14 : 499–509. [CrossRef] [PubMed] [Google Scholar]
- Till BG, Jensen MC, Wang J, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood 2012 ; 119 : 3940–50. [CrossRef] [PubMed] [Google Scholar]
- Ramos CA, Rouce R, Robertson CS, et al. In Vivo Fate and Activity of Second-versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol Ther 2018 ; 26 : 2727–37. [Google Scholar]
- Schubert M-L, Schmitt A, Hückelhoven-Krauss A, et al. Treatment of adult ALL patients with third-generation CD19-directed CAR T cells: results of a pivotal trial. J Hematol Oncol 2023 ; 16 : 79. [CrossRef] [PubMed] [Google Scholar]
- Enblad G, Karlsson H, Gammelgård G, et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin Cancer Res 2018 ; 24 : 6185–94. [CrossRef] [PubMed] [Google Scholar]
- Tomasik J, Jasiński M, Basak GW. Next generations of CAR-T cells - new therapeutic opportunities in hematology? Front Immunol 2022 ; 13 : 1034707. [CrossRef] [PubMed] [Google Scholar]
- Glienke W, Dragon AC, Zimmermann K, et al. GMP-Compliant Manufacturing of TRUCKs: CAR T Cells targeting GD2 and Releasing Inducible IL-18. Front Immunol 2022 ; 13 : 839783. [CrossRef] [PubMed] [Google Scholar]
- Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther 2015 ; 15 : 1145–54. [CrossRef] [PubMed] [Google Scholar]
- Tokarew N, Ogonek J, Endres S, et al. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer 2019 ; 120 : 26–37. [CrossRef] [PubMed] [Google Scholar]
- Uscanga-Palomeque AC, Chávez-Escamilla AK, Alvizo-Báez CA, et al. CAR-T Cell Therapy: From the Shop to Cancer Therapy. Int J Mol Sci 2023 ; 24 : 15688. [CrossRef] [PubMed] [Google Scholar]
- Zhang P, Zhang G, Wan X. Challenges and new technologies in adoptive cell therapy. J Hematol Oncol 2023 ; 16 : 97. [CrossRef] [PubMed] [Google Scholar]
- Zhao J, Lin Q, Song Y, et al. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol 2018 ; 11 : 132. [CrossRef] [PubMed] [Google Scholar]
- Lonez C, Breman E. Allogeneic CAR-T Therapy Technologies: Has the Promise Been Met? Cells 2024 ; 13 : 146. [CrossRef] [PubMed] [Google Scholar]
- Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017 ; 543 : 113–7. [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Jayaraman J, Mellody MP, Hou AJ, et al. CAR-T design: Elements and their synergistic function. EBioMedicine 2020 ; 58 : 102931. [CrossRef] [PubMed] [Google Scholar]
- Lamers CHJ, Sleijfer S, Vulto AG, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 2006 ; 24 : e20–2. [CrossRef] [PubMed] [Google Scholar]
- Lamers CH, Sleijfer S, Steenbergen S van, et al. Treatment of Metastatic Renal Cell Carcinoma With CAIX CAR-engineered T cells: Clinical Evaluation and Management of On-target Toxicity. Mol Ther 2013 ; 21 : 904. [Google Scholar]
- Daei Sorkhabi A, Mohamed Khosroshahi L, Sarkesh A, et al. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front Immunol. 2023 ; 14 : 1113882. [CrossRef] [Google Scholar]
- Mackensen A, Müller F, Mougiakakos D, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med 2022 ; 28 : 2124–32. [Google Scholar]
- Müller F, Taubmann J, Bucci L, et al. CD19 CAR T-Cell Therapy in Autoimmune Disease - A Case Series with Follow-up. N Engl J Med 2024 ; 390 : 687–700. [CrossRef] [PubMed] [Google Scholar]
- Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 2016 ; 127 : 3321–30. [CrossRef] [PubMed] [Google Scholar]
- Kadauke S, Myers RM, Li Y, et al. Risk-Adapted Preemptive Tocilizumab to Prevent Severe Cytokine Release Syndrome After CTL019 for Pediatric B-Cell Acute Lymphoblastic Leukemia: A Prospective Clinical Trial. J Clin Oncol 2021 ; 39 : 920–30. [CrossRef] [PubMed] [Google Scholar]
- Gust J, Hay KA, Hanafi L-A, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov 2017 ; 7 : 1404–19. [CrossRef] [PubMed] [Google Scholar]
- Hu Y, Sun J, Wu Z, et al. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J Hematol Oncol 2016 ; 9 : 70. [CrossRef] [PubMed] [Google Scholar]
- Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther J 2010 ; 18 : 843–51. [CrossRef] [Google Scholar]
- Orlando EJ, Han X, Tribouley C, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med 2018 ; 24 : 1504–6. [CrossRef] [PubMed] [Google Scholar]
- Kankeu Fonkoua LA, Sirpilla O, Sakemura R, et al. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol Ther - Oncolytics 2022 ; 25 : 69–77. [CrossRef] [Google Scholar]
Liste des tableaux
Exemples d’essais cliniques portant sur les cellules CAR-T (Clinicaltrials.gov). BCMA : antigène de maturation des cellules B (B-cell maturation antigen) ; CEA : antigène carcinoembryonnaire (carcinoembryonic antigen) ; GFRα4 : récepteur alpha-4 de la famille GDNF (GDNF family receptor Dα), ICAM/CD54 : molécule d’adhérence intercellulaire 1 (intercellular adhesion molecule 1). * : République populaire de Chine.
Liste des figures
|
Figure 1. Structure des CAR. Les CAR sont généralement composés de trois domaines : un domaine extracellulaire permettant de reconnaître un antigène à la surface des cellules tumorales ; un domaine transmembranaire pour assurer sa stabilité et son expression à la surface des lymphocytes T ; et un domaine intracellulaire de signalisation permettant d’émettre des signaux de co-stimulation et d’activer les lymphocytes T. |
| Dans le texte | |
|
Figure 2. Les générations de CAR. La reconnaissance de la cible tumorale est en général assurée par un scFv (bien que le même rôle puisse être joué par d’autres domaines de liaison). Le rectangle rouge représente le domaine d’activation (dérivé de la protéine CD3zeta [CD3ζ]) ; l’ovale bleu et le losange vert représentent les domaines de co-stimulation. L’étoile rose représente l’inducteur moléculaire, la flèche et le carré bleu clair symbolisent l’expression d’un gène, et les points bleu clair des cytokines qui peuvent être sécrétées. Le triangle jaune est le domaine IL-2Rβ nécessaire à l’activation de la voie JAK-STAT3/5. |
| Dans le texte | |
|
Figure 3. Quelques exemples des nouvelles générations de CAR. Parmi les CAR de nouvelle génération, certains sont capables de reconnaître deux ou plusieurs antigènes tumoraux distincts, soit par l’expression simultanée de plusieurs CAR, soit par l’utilisation de séquences de liaison ciblant plusieurs antigènes (porte logique « OR »). Les « split CAR » consistent à séparer sur deux CAR, le domaine d’activation et le domaine de signalisation, pour la reconnaissance de deux antigènes nécessaires à l’activation optimale des cellules CAR -T (porte logique « AND »). Dans les « SynNotch CAR », il est nécessaire que le récepteur Notch soit engagé lors de la reconnaissance de l’antigène (signal scFv 1) pour la transcription intracellulaire d’un CAR fonctionnel. Les cellules « Universal CAR » ont pour but d’assurer la polyvalence de la reconnaissance des antigènes tumoraux par un système intermédiaire. Ce système intermédiaire consiste en la production d’un CAR universel avec un scFv qui reconnaîtra une étiquette fusionnée à un scFv spécifique de différents antigènes tumoraux. Dans le cas du « split Universal Programmable CAR », le CAR est séparé en zipCAR (domaine de costimulation et d’activation) et zipFv (scFv pour la reconnaissance de l’antigène) liés par complémentarité pour former un CAR fonctionnel. Le système de « Small molecule protease-based regulation (SWIFF) CAR » est composé d’un CAR fusionné à son extrémité C-terminale à un site cible de protéase, d’une protéase et d’un composant de dégradation des protéines (degron). La protéase est active dans des conditions normales avec la production d’un CAR fonctionnel. Lors de l’administration d’un inhibiteur de la protéase (par exemple l’asunaprevir), le degron est conservé, ce qui entraîne la dégradation du CAR. Dans le système « inhibitor CAR », le CAR est exprimé simultanément avec un autre récepteur contenant un signal inhibiteur qui reconnaît les tissus sains et empêche l’activité de la cellule CAR-T. Un autre système de sécurité dans le CAR est l’incorporation de gènes ou de commutateurs de suicide contrôlables (par exemple, iCasp9), coexprimés avec le CAR pour tuer les cellules CAR-T en cas d’effets secondaires graves. |
| Dans le texte | |
|
Figure 4. Mode d’action d’une cellule CAR-T. La partie extracellulaire du CAR se lie spécifiquement à l’antigène exprimé à la surface des cellules tumorales, ce qui active le recrutement de la protéine ZAP70 par le domaine d’activation (CD3ζ) et le déclenchement d’une cascade d’activation. Le domaine de co-stimulation permet d’activer d’autres cascades de signalisation (PI3K dans le cas de CD28, et TRAF1/2 dans le cas de 4-1BB). Ces cascades agissent sur les voies contrôlées par NF-κB et STAT3, induisant par exemple la production de cytokines telles que l’IL-2, l’IL-5 et l’IFNγ. L’ensemble de ces réactions favorisent l’expansion et la toxicité des lymphocytes T, notamment par la libération de perforine et de granzyme. L’introduction de granzyme dans la cellule tumorale, via les pores formés par la perforine, induit la mort de la cellule. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.