")
")
| Issue |
Med Sci (Paris)
Volume 31, Number 11, Novembre 2015
|
|
|---|---|---|
| Page(s) | 989 - 995 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20153111013 | |
| Published online | 17 novembre 2015 | |
L’angiogenèse tumorale
Quand l’arbre de vie tourne mal
Tumor angiogenesis: when the Tree of Life turns bad
1
Institut Cochin, CNRS UMR8104, Inserm U1016, université Paris Descartes, 22, rue Méchain, 75014
Paris, France
2
Centre de recherche en cancérologie Nantes-Angers, CNRS UMR6299, Inserm U892, université de Nantes, 8, quai Moncousu, 44000
Nantes, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
; julie.gavard@LabSoap
Résumé
L’angiogenèse correspond à la formation de nouveaux vaisseaux sanguins au cours du développement et chez l’adulte. Les mécanismes moléculaires et cellulaires de ce processus sont piratés au cours de l’angiogenèse tumorale, c’est-à-dire lors de la mise en place d’une vascularisation dédiée à l’irrigation du tissu cancéreux. Cependant, les processus déployés sont particulièrement inefficaces dans un environnement hostile, conduisant à un réseau anormal. Cette synthèse présente un état des connaissances de la formation normale et pathologique des vaisseaux sanguins et décrit comment les aberrations du réseau vasculaire tumoral contribuent à la tumorigenèse et aux échecs de thérapies conventionnelles.
Abstract
Angiogenesis is a highly controlled multistep process that allows the formation of a harmonious vascular network during embryonic development and in adults. In addition, tumor progression also involves a dedicated blood vessel supply to fuel the tumor mass, pirating physiological molecular and cellular mechanisms. However, tumor angiogenesis is a quite inefficient process, as perfusion is not optimal, vessel integrity is not guaranteed, and vessel network is irrationally organized. While fundamental molecular and cellular mechanisms have been the field of intense investigation, anticancer therapies have evolved with the possibility to target tumor angiogenesis. This review presents the different steps involved in the formation of normal and tumor angiogenesis, and how tumor vasculature abnormalities could contribute to tumorigenesis and conventional therapy failure.
© 2015 médecine/sciences – Inserm
Par une approche empirique classique, révolutionnée par l’avènement du microscope, Marcello Malpighi fut le premier à mettre à jour en 1661 l’existence d’un réseau capillaire dans les poumons de grenouille. Tandis que les connaissances sur l’anatomie de la circulation sanguine et l’irrigation dans les organismes entiers vivants ont progressé au fil des siècles, il a fallu attendre les années 1960-1970 et l’apparition des premières cultures de cellules endothéliales pour que les processus d’angiogenèse commencent à être étudiés et que leur implication dans les tumeurs soit proposée. Dès lors, les mécanismes cellulaires et moléculaires mis en jeu n’ont cessé d’être modélisés et déchiffrés jusqu’à en devenir la cible de thérapies, notamment anticancéreuses.
L’angiogenèse correspond à la formation de nouveaux vaisseaux depuis un réseau préexistant, différant ainsi de la vasculogenèse qui est un processus essentiellement développemental au cours duquel un vaisseau s’établit de novo à partir de précurseurs cellulaires [1]. Parmi les différents modes opératoires employés par l’angiogenèse, nous décrirons ici le bourgeonnement, dont les mécanismes moléculaires sont les plus largement connus. Chez l’adulte, la majorité des cellules endothéliales demeurent dans un état de quiescence. L’angiogenèse n’est normalement observée que dans des conditions ou au sein de mécanismes physiologiques strictement régulés comme la réparation tissulaire, le cycle menstruel, la grossesse ou pour répondre à des besoins particuliers en oxygène et en nutriments. Une altération de ces processus peut engendrer des anomalies de l’homéostasie vasculaire et du tissu irrigué avec, souvent, des conséquences sur les grandes fonctions organiques. Dans cette revue, nous décrirons les mécanismes de l’angiogenèse normale, les anomalies caractéristiques de l’angiogenèse tumorale et les stratégies thérapeutiques anticancéreuses ciblant la vascularisation tumorale.
L’angiogenèse : une valse à quatre temps
Premier temps : la sélection
La première étape de l’angiogenèse correspond à l’activation et à la sélection de cellules endothéliales, interchangeables quant à leur fonction, qui conditionnent le déroulement de l’élongation [2]. Bien que plus de 99 % des cellules endothéliales soient quiescentes, un état qui leur est imposé notamment par les voies de signalisation BMP (bone morphogenetic proteins) et FGF-2 (fibroblast growth factor 2), ces cellules n’en demeurent pas moins extrêmement plastiques et réactives aux facteurs environnementaux. Parmi ces facteurs, la balance entre les signaux pro- et anti-angiogéniques gouverne le devenir des cellules endothéliales, pouvant alors les entraîner rapidement vers un phénotype angiogénique et l’acquisition de propriétés migratoires ou prolifératives uniques (Figure 1A). Ces deux comportements les répartissent (1) en cellules de front (tip cell) qui bourgeonnent à partir du vaisseau existant, se dirigent vers la source du signal angiogénique et guident le néo-vaisseau ; (2) et en cellules de soutien (stalk cells) qui prolifèrent en arrière, permettant l’allongement du vaisseau. D’un point de vue moléculaire, deux voies de signalisation, reposant sur le DLL4 (delta-like ligand 4), un ligand de Notch, et le VEGF (vascular endothelial growth factor) et son récepteur VEGF-R (VEGF receptor), sont largement impliquées dans le processus de sélection de la cellule de front [2]. En effet, une concentration optimale en VEGF induit, en interagissant avec son récepteur VEGF-R2, l’augmentation de la transcription du gène codant DLL4 dans la cellule de front [3] (Figure 2). La protéine DLL4 interagit alors avec son récepteur Notch1 dans les cellules endothéliales situées au voisinage [4]. L’activation de cette voie dans les cellules de soutien diminue l’expression du gène VEGF-R2 et augmente celle de VEGF-R1, par l’intermédiaire des facteurs de transcription Hes et Hey1 [3–5]. Bien que dépourvu d’activité transductionnelle, VEGF-R1 s’oppose à la signalisation induite par VEGF-R2 par compétition et séquestration de leur ligand commun, le VEGF. Ce mécanisme permet que seule la cellule de front, qui exprime fortement VEGF-R2 et la protéine DLL4, puisse répondre au signal pro-angiogénique induit par le VEGF. De manière intéressante, cette compétition de signalisation (DLL4 versus VEGF), et donc de positionnement de la cellule de front, pourrait assurer une sélection continue durant l’allongement du vaisseau et intégrer d’autres signaux morphogénétiques tels que ceux induits par les BMP responsables de la quiescence des cellules [5].
 |
Figure 1. Les étapes de l’angiogenèse normale. A. Une concentration adéquate en VEGF (vascular endothelial growth factor) permet l’expression du DLL4 (delta-like ligand 4) dans la cellule de front et l’activation de la voie de signalisation impliquant son récepteur, Notch, dans les cellules de soutien. La matrice extracellulaire est dégradée localement et les interactions péricytes-cellules endothéliales sont modulées. B. En réponse aux facteurs environnants, les jonctions endothéliales de la cellule de front sont affaiblies, acquièrent un phénotype invasif et interagissent avec la matrice extracellulaire. C. Les cellules de soutien prolifèrent et permettent la croissance et la rencontre de deux bourgeonnements vasculaires. Les processus de vacuolisation commencent. D. La fusion des vacuoles endothéliales conduit à la formation de la lumière et permet la perfusion du nouveau vaisseau. Finalement, une nouvelle membrane basale est produite et les péricytes nouvellement recrutés stabilisent les jonctions endothéliales. |
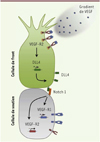 |
Figure 2. Les mécanismes moléculaires de sélection de la cellule de front. Un gradient de VEGF (vascular endothelial growth factor) permet d’activer son récepteur VEGF-R2 et d’induire la transcription de DLL4 (delta-like ligand 4) dans la cellule de front. Ligand de Notch, DLL4 active cette voie de signalisation dans les cellules de soutien à proximité, ce qui conduit à l’expression du récepteur VEGF-R1 et à la répression de la transcription du gène codant le VEGF-R2. Le VEGF-R1, dépourvu d’activité transductionnelle, séquestre le VEGF libre et prévient l’activation de VEGF-R2. |
Deuxième temps : la migration
À la suite de son activation et de sa sélection, la cellule de front bourgeonne à partir du vaisseau et acquiert de nombreuses protrusions membranaires de type filopodes qui sont hautement dynamiques [1, 2] (Figure 1B). Dans le modèle du poisson zèbre, l’analyse des vaisseaux intersomitiques montre que ces filopodes sont impliqués dans la vitesse de migration mais qu’ils ne participent pas à l’orientation de la cellule [7]. L’acquisition de cette capacité de migration repose sur un affaiblissement des interactions entre cellules endothéliales et une dégradation de la matrice extracellulaire, tout en maintenant une certaine intégrité du vaisseau. La perméabilité vasculaire est donc momentanément augmentée et contribue, en retour, à la diffusion de facteurs moléculaires et cellulaires pro-angiogéniques [1]. La stimulation des cellules endothéliales induit, en outre, le démantèlement des jonctions adhérentes notamment via l’endocytose de la cadhérine endothéliale (VE-cadherin [vascular endothelial-cadherin]) [8–10]. En fonction de ces observations, les travaux de l’équipe d’Holger Gerhardt ont récemment permis d’expliquer le déroulement et la dynamique de la migration endothéliale. Ainsi, les taux élevés de VEGF que la cellule de front reçoit, induisent une internalisation de la cadhérine endothéliale, favorisant ainsi la migration de la cellule, tandis que l’activation de la voie de signalisation dépendant de Notch, induite par le VEGF, inhibe les protrusions membranaires et favorise l’adhérence cellulaire permettant une migration collective en arrière [11]. Le groupe de Peter Carmeliet a mis à jour un lien direct entre la migration endothéliale et le métabolisme énergétique. En effet, la déplétion in vitro et in vivo de la phosphatase PFKFB3 (phosphofructokinase-2/fructose-2,6-bisphosphatase), une enzyme clé de la glycolyse, inhibe la migration des cellules de front et la prolifération des cellules de soutien [12]. De plus, l’expression de la PFKFB3 est augmentée par des signaux pro-angiogéniques de sélection des cellules de front (VEGF et hypoxie, par exemple). Inversement, elle est réduite en présence de la DLL4 [12]. Enfin, la phosphatase PFKFB3 s’accumule dans les filopodes des cellules de front où elle interagit avec le réseau d’actine filamenteuse. Cette localisation pourrait permettre de garantir un accès facilité à une grande quantité d’ATP qui est nécessaire pour le remodelage membranaire et pour l’acquisition du phénotype migratoire de la cellule.
Troisième temps : la fusion et la formation de la lumière
Sous l’impulsion de la prolifération des cellules de soutien, le néo-vaisseau s’allonge, guidé par la cellule de front. Ce processus repose sur l’établissement d’un gradient de VEGF-C qui est sécrété par les macrophages présents au niveau des sites de bourgeonnement. Par l’intermédiaire de son récepteur VEGF-R3, le VEGF-C stimule les cibles transcriptionnelles de la voie de signalisation impliquant Notch et diminue la sensibilité des cellules de front au VEGF [6]. Lorsque deux cellules de front entrent en contact au travers de leurs filopodes, les deux bourgeons vasculaires fusionnent par anastomose et forment un vaisseau connecté [2] (Figure 1C). Une drogue bloquant le cytosquelette d’actine inhibe la formation de cette connexion [7]. Cette interaction entraîne, de manière concomitante, la formation de la lumière du vaisseau. Les mécanismes impliqués dans ce processus ne sont pas entièrement élucidés, notamment à cause de la difficulté à développer des modèles expérimentaux permettant d’étudier spécifiquement cette étape. Néanmoins, la formation et la fusion de larges vacuoles dans les cellules endothéliales pourraient participer à l’établissement de la lumière, en coopération avec la luménisation des cellules de front et la force de pression exercée par la circulation sanguine [13]. La vacuolisation des cellules repose sur leurs interactions avec la matrice extracellulaire. En effet, dans un modèle murin, l’absence, dans le compartiment endothélial, de l’intégrine α1 qui se lie à la matrice, n’est pas viable et les embryons des souris présentent de multiples obstructions vasculaires [14].
Quatrième temps : la maturation et le flux sanguin
In fine, la maturation du vaisseau nécessite le rétablissement de la barrière endothéliale et du flux sanguin (Figure 1D). La voie de signalisation impliquant l’angiopoïétine 1 (Ang1) et son récepteur Tie2 s’oppose aux actions du VEGF. Elle induit le renforcement des jonctions entre les cellules endothéliales ainsi que leur quiescence [15, 16]. Un gradient de PDGF-BB (platelet-derived growth factor) et de TGF-β1 (transforming growth factor) induit parallèlement le recrutement des péricytes1 et des cellules musculaires lisses, ainsi que le dépôt d’une nouvelle matrice extracellulaire à la surface des vaisseaux, assurant ainsi la maturation finale d’un vaisseau sanguin fonctionnel.
À tout moment du processus, sous l’effet de modifications du microenvironnement ou d’adaptations physiologiques, l’angiogenèse peut être arrêtée voire inversée [17]. Les bourgeons vasculaires sont alors éliminés par rétraction (pruning) ou apoptose, montrant la plasticité et la dynamique des événements morphogénétiques impliqués, et donc la nécessité d’un contrôle strict pour le bon fonctionnement de l’angiogenèse. Ces observations rendent ainsi compte du degré de complexité et de plasticité de l’angiogenèse.
L’angiogenèse tumorale : une valse à mille temps
En 1963, les travaux pionniers de Judah Folkman ont apporté la preuve expérimentale que la croissance tumorale requérait une néo-vascularisation qui lui était dédiée [18]. En effet, il a observé que l’implantation de cellules de mélanome de souris dans la thyroïde de chien cultivée ex vivo, conduisait à la formation de petites tumeurs qui ne dépassaient pas 1 à 2 mm de diamètre et qui étaient dépourvues de vaisseaux sanguins. Au contraire, lorsque ces cellules cancéreuses étaient introduites dans des souris syngéniques, les tumeurs étaient vascularisées et se développaient rapidement. Ces travaux pionniers ont donné naissance à de nombreuses études visant à mieux comprendre les mécanismes de la vascularisation tumorale et, par conséquent, à la recherche de stratégies thérapeutiques afin de normaliser le réseau vasculaire tumoral.
Les anomalies de l’angiogenèse tumorale
La formation d’un réseau vasculaire dédié à l’apport en oxygène et en nutriments fait partie intégrante de la signature tumorale [19]. Ce réseau correspond à la fois à l’exploitation détournée des mécanismes physiologiques fondamentaux, en particulier ceux décrits ci-dessus participant à l’angiogenèse développementale, mais aussi à des processus inédits, souvent peu élucidés, voire encore débattus dans la communauté scientifique [20]. Nous les présentons brièvement (Figure 3).
 |
Figure 3. Les différents modes de formation des vaisseaux tumoraux. 1. L’angiogenèse par bourgeonnement qui dépend des gradients en VEGF conduisant à la sélection d’une ou plusieurs cellules de front. 2. Le recrutement de novo de précurseurs endothéliaux issus de la moelle osseuse. 3. Le mimétisme vasculaire des cellules tumorales et l’incorporation directe de vaisseaux sanguins. 4. La transdifférenciation endothéliale de cellules tumorales à caractère souche. 5. La séparation d’un vaisseau sanguin par intussusception qui conduit à l’accroissement de la ramification des vaisseaux tumoraux. VEGF : vascular endothelial growth factor. |
-
L’angiogenèse par bourgeonnement (qui est le processus le plus étudié) dépend particulièrement des concentrations en VEGF dans le milieu ; le nombre de cellules de front sélectionné et le nombre de filopodes apparaissent anormalement élevés.
-
Des cellules endothéliales précurseurs, circulantes ou résidantes de la moelle osseuse ou des parois vasculaires, pourraient être recrutées au sein de la tumeur et établir un réseau sanguin de novo par vasculogenèse.
-
Le mimétisme vasculaire, qui correspond à la capacité de cellules tumorales d’intégrer directement les vaisseaux sanguins et d’augmenter artificiellement et de manière aberrante la masse vasculaire, a été mis en évidence dans des tumeurs hautement agressives comme les mélanomes métastatiques.
-
Un autre mode de vascularisation tumorale relève directement des propriétés des cellules tumorales. Il implique la transdifférenciation de cellules cancéreuses non différenciées qui présentent un caractère de cellule souche, comme cela a été observé dans les glioblastomes [1].
-
La séparation en deux d’un vaisseau sanguin pré-existant par intussusception2 participerait à la ramification et à l’architecture chaotique des vaisseaux tumoraux.
Quelle que soit l’importance du processus de vascularisation déployé pour irriguer la masse tumorale, celui-ci qui est généralement peu efficace et désorganisé, mais contribue néanmoins à l’agressivité de la tumeur et à son éventuelle dissémination.
Structure et caractéristiques des vaisseaux tumoraux
Une perméabilité constitutive importante, une dilatation, un réseau chaotique et un flux de sang aberrant caractérisent les vaisseaux tumoraux (Figure 4). Au sein d’un même vaisseau, le flux sanguin n’est pas constant et peut même changer de direction [1]. Du fait de la perméabilité vasculaire, la pression interstitielle est haute. Les phénomènes d’œdème, de fibrose et d’inflammation sont ainsi exacerbés. L’angiogenèse et la dissémination tumorale sont par là même favorisées [21].
 |
Figure 4. Les aberrations du réseau vasculaire tumoral. L’organisation générale d’un réseau vasculaire normal structuré et hiérarchisé, s’oppose à celle de la vascularisation aberrante et tortueuse observée dans les tumeurs. Les vaisseaux ont une perméabilité vasculaire anormale associée à une déstabilisation des protéines de jonctions comme la cadhérine endothéliale, une membrane basale discontinue, des péricytes peu nombreux et lâches, un flux sanguin inconstant et de multiples anastomoses vasculaires. |
Les vaisseaux tumoraux ne forment qu’une barrière partielle contre les grosses molécules et les protéines plasmatiques. Ainsi, on peut montrer dans des expériences d’extravasation de Dextran3 de différentes masses moléculaires, qui permettent de mesurer la perméabilité vasculaire, que les molécules de petite taille pénètrent plus profondément les tissus tumoraux mais qu’elles sont rapidement éliminées. En revanche les molécules de taille plus importante (de 40-70 kDa) s’accumulent préférentiellement à la surface des vaisseaux où elles semblent persister [22]. Outre un défaut de recouvrement par les péricytes et une membrane basale discontinue, les jonctions endothéliales des vaisseaux tumoraux sont également aberrantes. Le nombre de cellules positives pour le marqueur des cellules musculaires lisses, α-SMA (smooth muscle actin), est en effet réduit dans les modèles de xénogreffes [23]. De même, dans des modèles ex vivo de glioblastomes, les jonctions formées par la cadhérine endothéliale sont désorganisées [9, 10]. Associée à un microenvironnement riche en facteurs pro-angiogéniques et inflammatoires, et aux aberrations touchant la composition des vaisseaux tumoraux, la perméabilité vasculaire est anormalement élevée au sein des tumeurs.
Ces fuites vasculaires provoquent une pression interstitielle importante et la formation d’œdèmes vasculaires, ce qui rend l’adressage et la délivrance des drogues difficiles et hétérogènes au sein de la tumeur [21].
La part de l’angiogenèse dans la progression tumorale
L’angiogenèse tumorale devient vite primordiale pour le développement de la tumeur. Les néo-vaisseaux tumoraux défectueux participent eux aussi à créer un environnement métabolique et immunitaire unique [24]. En effet, l’angiogenèse tumorale participe à l’augmentation de l’afflux en oxygène et en nutriments, dont l’apport par simple diffusion n’est plus suffisant à la croissance de la tumeur (l’oxygène ne peut diffuser passivement que sur de courtes distances, de l’ordre d’une centaine de micromètres). L’environnement tumoral est donc enrichi en nombreux facteurs pro-angiogéniques, par exemple, en réponse à l’activation des voies d’hypoxie, au microenvironnement inflammatoire ou encore aux aberrations métaboliques ou génétiques que présentent les cellules tumorales [20, 25].
En plus de la croissance tumorale, les conditions développées dans l’environnement des tumeurs exacerbent leur agressivité. Ceci est réalisé par la sélection au sein de la masse tumorale des cellules les plus pro-angiogéniques, par la dissémination métastasique, l’acquisition de chimio- et de radio-résistance, l’échappement immunitaire, etc. [20, 25]. La découverte récente de cellules tumorales à caractère souche suggère, de plus, l’existence d’une niche vasculaire tumorale [26]. En effet, des cellules souches cancéreuses ont été observées dans le voisinage direct des cellules endothéliales. Elles pourraient contribuer au maintien de cette sous-population tumorale aux propriétés uniques [26, 27].
Dans ce contexte, les efforts en recherche anticancéreuse ont conduit pendant les dernières décennies au développement de molécules visant à bloquer l’angiogenèse tumorale afin d’affamer les tumeurs et/ou à bloquer les voies de sortie métastatique. En particulier, le bevacizumab, un anticorps bloquant le VEGF, s’est avéré efficace dans les cancers métastatiques colorectaux et rénaux en combinaison avec une chimiothérapie [20]. Son utilisation reste encore débattue en ce qui concerne les glioblastomes et les cancers du sein. Bien qu’ayant une action anti-tumorale réelle, le bevacizumab pourrait favoriser l’acquisition d’un phénotype plus agressif et la dissémination métastatique, comme cela a été montré dans des modèles expérimentaux de glioblastomes et de cancers du sein et du pancréas [28–30]. Des mécanismes de résistance aux anti-VEGF suggèrent que d’autres molécules pro-angiogéniques sont impliquées dans le processus de développement tumoral, ou que les cellules endothéliales tumorales sont devenues insensibles au VEGF pour leur croissance.
Ces traitements anti-angiogéniques permettent de normaliser la vasculature tumorale, c’est-à-dire de rétablir la structure et le fonctionnement physiologique du réseau sanguin tumoral [30]. La stratégie de restauration des vaisseaux pourrait permettre une réduction de la pression interstitielle et une diminution des œdèmes tout en permettant la perfusion et l’oxygénation des tumeurs. Cela pourrait alors faciliter l’adressage d’agents chimiothérapiques, sensibiliser la masse tumorale aux radiothérapies ou encore permettre d’assister les immunothérapies [30].
Dans ce contexte, plusieurs stratégies sont envisagées et examinées. Elles comprennent le rétablissement de la balance entre les facteurs pro- et anti-angiogéniques, la restauration de la barrière endothéliale en ciblant les jonctions entre cellules endothéliales et entre cellules endothéliales et péricytes, ou l’activation des voies de maturation des vaisseaux.
La formation d’un réseau sanguin dédié à l’irrigation du tissu tumoral est donc un processus perverti et inefficace qui détourne et adapte les mécanismes physiologiques de l’angiogenèse. Les découvertes de molécules anti-angiogéniques et leurs applications cliniques ont révélé que la seule abolition des vaisseaux tumoraux ne pourrait suffire à diminuer la croissance tumorale et à réduire sa dissémination. Ainsi, il semblerait qu’au-delà du blocage de l’angiogenèse tumorale, la normalisation des vaisseaux tumoraux pourrait être une stratégie prometteuse. Dans cette optique, il est nécessaire, en tenant compte de leur microenvironnement, de considérer les cellules endothéliales comme des cibles potentielles.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Les auteurs remercient l’ensemble du laboratoire Signalisation en oncogenèse, angiogenèse et perméabilité (@LabSoap). Les recherches reçoivent le support de la Ligue nationale contre le cancer, l’INCA, la Fondation pour la recherche médicale, la Région Pays-de-La-Loire, et Nantes-Métropole. Lucas Treps est lauréat d’un contrat doctoral de l’université Paris Descartes.
Les péricytes sont des cellules qui entourent les vaisseaux et peuvent jouer le rôle de cellules mésenchymateuses.
Invagination.
Le dextran est un ensemble de polysaccharides à longue chaîne, de masse moléculaire variable utilisé pour mesurer la perméabilité de différentes structures tissulaires.
Références
- Azzi S, Gavard J. Vaisseaux sanguins et tumeurs ou l’art du dialogue. Med Sci (Paris) 2014 ; 30 : 408–414. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Wacker A, Gerhardt H. Endothelial development taking shape. Curr Opin Cell Biol 2011 ; 23 : 676–685. [PubMed] [Google Scholar]
- Hellstrom M, Phng LK, Hofmann JJ, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007 ; 445 : 776–780. [CrossRef] [PubMed] [Google Scholar]
- Suchting S, Freitas C, le Noble F, et al. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci USA 2007 ; 104 : 3225–3230. [CrossRef] [Google Scholar]
- Jakobsson L, Franco CA, Bentley K, et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 2010 ; 12 : 943–953. [Google Scholar]
- Tammela T, Zarkada G, Wallgard E, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008 ; 454 : 656–660. [CrossRef] [PubMed] [Google Scholar]
- Phng LK, Stanchi F, Gerhardt H. Filopodia are dispensable for endothelial tip cell guidance. Development 2013 ; 140 : 4031–4040. [CrossRef] [PubMed] [Google Scholar]
- Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 2006 ; 8 : 1223–1234. [CrossRef] [PubMed] [Google Scholar]
- Le Guelte A, Galan-Moya EM, Dwyer J, et al. Semaphorin 3A elevates endothelial cell permeability through PP2A inactivation. J Cell Sci 2012 ; 125 : 4137–4146. [CrossRef] [PubMed] [Google Scholar]
- Dwyer J, Hebda JK, Le Guelte A, et al. Glioblastoma cell-secreted interleukin-8 induces brain endothelial cell permeability via CXCR2. PLoS One 2012 ; 7 : e45562. [CrossRef] [PubMed] [Google Scholar]
- Bentley K, Franco CA, Philippides A, et al. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol 2014 ; 16 : 309–321. [CrossRef] [PubMed] [Google Scholar]
- De Bock K, Georgiadou M, Schoors S, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013 ; 154 : 651–663. [CrossRef] [PubMed] [Google Scholar]
- Iruela-Arispe ML, Davis GE. Cellular and molecular mechanisms of vascular lumen formation. Dev Cell 2009 ; 16 : 222–231. [CrossRef] [PubMed] [Google Scholar]
- Zovein AC, Luque A, Turlo KA, et al. Beta1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev Cell 2010 ; 18 : 39–51. [CrossRef] [PubMed] [Google Scholar]
- Gavard J, Patel V, Gutkind JS. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell 2008 ; 14 : 25–36. [CrossRef] [PubMed] [Google Scholar]
- Fukuhara S, Sako K, Minami T, et al. Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat Cell Biol 2008 ; 10 : 513–526. [CrossRef] [PubMed] [Google Scholar]
- Ricard N, Simons M. When it is better to regress: dynamics of vascular pruning. PLoS Biol 2015 ; 13 : e1002148. [CrossRef] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis. Annu Rev Med 2006 ; 57 : 1–18. [CrossRef] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011 ; 144 : 646–674. [CrossRef] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011 ; 473 : 298–307. [CrossRef] [PubMed] [Google Scholar]
- Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Front Oncol 2013 ; 3 : 211. [CrossRef] [PubMed] [Google Scholar]
- Dreher MR, Liu W, Michelich CR, et al. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst 2006 ; 98 : 335–344. [CrossRef] [PubMed] [Google Scholar]
- Inai T, Mancuso M, Hashizume H, et al. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol 2004 ; 165 : 35–52. [CrossRef] [PubMed] [Google Scholar]
- Sapieha P, Zaniolo K, Hamel D, et al. L’offre et la demande : l’influence du métabolisme énergétique sur l’angiogenèse. Med Sci (Paris) 2009 ; 25 : 346–348. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Gothie E, Pouyssegur J. HIF-1: régulateur central de l’hypoxie. Med Sci (Paris) 2002 ; 18 : 70–78. [CrossRef] [EDP Sciences] [Google Scholar]
- Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007 ; 11 : 69–82. [CrossRef] [PubMed] [Google Scholar]
- Galan-Moya EM, Le Guelte A, Fernandes EL, et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep 2011 ; 12 : 470–476. [CrossRef] [PubMed] [Google Scholar]
- Paez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009 ; 15 : 220–231. [Google Scholar]
- Ebos JM, Lee CR, Cruz-Munoz W, et al. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009 ; 15 : 232–239. [CrossRef] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011 ; 10 : 417–427. [Google Scholar]
Liste des figures
|
Figure 1. Les étapes de l’angiogenèse normale. A. Une concentration adéquate en VEGF (vascular endothelial growth factor) permet l’expression du DLL4 (delta-like ligand 4) dans la cellule de front et l’activation de la voie de signalisation impliquant son récepteur, Notch, dans les cellules de soutien. La matrice extracellulaire est dégradée localement et les interactions péricytes-cellules endothéliales sont modulées. B. En réponse aux facteurs environnants, les jonctions endothéliales de la cellule de front sont affaiblies, acquièrent un phénotype invasif et interagissent avec la matrice extracellulaire. C. Les cellules de soutien prolifèrent et permettent la croissance et la rencontre de deux bourgeonnements vasculaires. Les processus de vacuolisation commencent. D. La fusion des vacuoles endothéliales conduit à la formation de la lumière et permet la perfusion du nouveau vaisseau. Finalement, une nouvelle membrane basale est produite et les péricytes nouvellement recrutés stabilisent les jonctions endothéliales. |
| Dans le texte | |
|
Figure 2. Les mécanismes moléculaires de sélection de la cellule de front. Un gradient de VEGF (vascular endothelial growth factor) permet d’activer son récepteur VEGF-R2 et d’induire la transcription de DLL4 (delta-like ligand 4) dans la cellule de front. Ligand de Notch, DLL4 active cette voie de signalisation dans les cellules de soutien à proximité, ce qui conduit à l’expression du récepteur VEGF-R1 et à la répression de la transcription du gène codant le VEGF-R2. Le VEGF-R1, dépourvu d’activité transductionnelle, séquestre le VEGF libre et prévient l’activation de VEGF-R2. |
| Dans le texte | |
|
Figure 3. Les différents modes de formation des vaisseaux tumoraux. 1. L’angiogenèse par bourgeonnement qui dépend des gradients en VEGF conduisant à la sélection d’une ou plusieurs cellules de front. 2. Le recrutement de novo de précurseurs endothéliaux issus de la moelle osseuse. 3. Le mimétisme vasculaire des cellules tumorales et l’incorporation directe de vaisseaux sanguins. 4. La transdifférenciation endothéliale de cellules tumorales à caractère souche. 5. La séparation d’un vaisseau sanguin par intussusception qui conduit à l’accroissement de la ramification des vaisseaux tumoraux. VEGF : vascular endothelial growth factor. |
| Dans le texte | |
|
Figure 4. Les aberrations du réseau vasculaire tumoral. L’organisation générale d’un réseau vasculaire normal structuré et hiérarchisé, s’oppose à celle de la vascularisation aberrante et tortueuse observée dans les tumeurs. Les vaisseaux ont une perméabilité vasculaire anormale associée à une déstabilisation des protéines de jonctions comme la cadhérine endothéliale, une membrane basale discontinue, des péricytes peu nombreux et lâches, un flux sanguin inconstant et de multiples anastomoses vasculaires. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.