")
")
| Issue |
Med Sci (Paris)
Volume 26, Number 6-7, Juin–Juillet 2010
|
|
|---|---|---|
| Page(s) | 627 - 635 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2010266-7627 | |
| Published online | 15 juin 2010 | |
Chaperons pharmacologiques
Un espoir thérapeutique pour les pathologies conformationnelles
Pharmacological chaperones : a potential therapeutic treatment for conformational diseases
Institut de génomique fonctionnelle, CNRS UMR 5203, Inserm U661, Universités Montpellier 1 et 2, 141, rue de la Cardonille, 34094 Montpellier Cedex 05, France
* Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
* Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
De nombreuses maladies génétiques ou neurodégénératives sont consécutives à un repliement défectueux de protéines entraînant leur dégradation ou leur accumulation sous forme d’agrégats. Ces maladies sont regroupées sous le terme de « pathologies conformationnelles ». Les protéines impliquées n’étant pas correctement repliées, elles sont disqualifiées par le système contrôle-qualité cellulaire. En conséquence, elles ne peuvent plus exercer leur rôle physiologique. Des composés spécifiques (ligands, substrats, inhibiteurs), appelés chaperons pharmacologiques, sont capables de se lier à ces protéines défectueuses, de les stabiliser afin de leur permettre d’acquérir une conformation quasi native, d’échapper au système contrôle-qualité et par conséquent de récupérer une fonctionnalité. Ces molécules ont une activité intrinsèque variable : elles peuvent être agonistes (activatrices), antagonistes (inhibitrices) ou modulateurs allostériques de leurs récepteurs, canaux ioniques ou enzymes cibles. Les chaperons pharmacologiques représentent donc un espoir thérapeutique pour des maladies rares comme la mucoviscidose, certaines rétinites pigmentaires, le diabète insipide néphrogénique, la maladie de Fabry ou de Gaucher, mais aussi pour certains cancers et des pathologies du système nerveux très invalidantes comme les maladies d’Alzheimer ou de Parkinson.
Abstract
Many genetic and neurodegenerative diseases in humans result from protein misfolding and/or aggregation. These diseases are named conformational diseases. As a result, the misfolded non functional proteins are rejected and misrouted by the cellular quality control system, and cannot play their endogenous physiological roles. Specific compounds (ligands, substrates or inhibitors) known as pharmacological chaperones are able to bind and stabilize these misfolded proteins. Their interaction allows the target proteins to escape the quality control system and to be functionally rescued. These pharmacochaperones may possess different intrinsic activity : they can be antagonists (inhibitors), agonists (activators) or allosteric modulators of the target receptors, ionic channels or enzymes. Pharmacological chaperones have obviously a therapeutic potential to treat rare diseases like cystic fibrosis, retinitis pigmentosa, nephrogenic diabetes insipidus, Fabry disease, Gaucher disease, but also for cancers and more frequent and highly invalidant neurodegenerative disorders such as Alzheimer’s disease or Parkinson’s disease.
© 2010 médecine/sciences - Inserm / SRMS
Repliement défectueux des protéines et pathologies associées
Pour qu’une protéine soit active, elle doit acquérir une structure tridimensionnelle unique dite « native » qui correspond à l’état énergétique le plus stable. Cette étape de repliement, qui dépend de la séquence en acides aminés de la protéine elle-même et de l’environnement cellulaire local, est cruciale. Elle peut s’effectuer de façon coordonnée à la synthèse de la protéine considérée. Le repliement des protéines prend place dans trois compartiments cellulaires : le cytoplasme, le réticulum endoplasmique (RE) et les mitochondries. Nous nous limiterons dans cette revue au processus de repliement qui intervient dans le RE, point de départ de la voie de sécrétion des protéines vers le milieu extracellulaire, de l’export des protéines vers la membrane plasmique ou du ciblage vers d’autres compartiments membranaires impliqués dans la sécrétion ou l’endocytose [1, 2]. Le RE est le siège d’un système contrôle-qualité des protéines qui fait intervenir de nombreux chaperons moléculaires et enzymes. Le rôle de ceux-ci est de reconnaître les protéines incomplètement ou mal repliées, de catalyser leur repliement efficace lors de plusieurs étapes par exemple l’établissement de ponts disulfures ou l’accrochage de chaînes glycosylées et de les assembler si nécessaire [1, 2]. Ces chaperons moléculaires sont endogènes à la cellule [3], par opposition aux chaperons chimiques ou pharmacologiques que nous définirons plus loin. Une fois synthétisées et correctement repliées, les protéines rejoignent le compartiment cellulaire dans lequel elles jouent leur rôle physiologique, qu’il s’agisse de la membrane pour des récepteurs ou des canaux ioniques, du lysosome pour de nombreuses enzymes du métabolisme, ou du milieu extracellulaire pour des protéines sécrétées (anticorps par exempl e). Si une protéine n’acquiert pas sa structure native malgré l’action du système contrôle-qualité, elle est dirigée vers le cytoplasme à travers le translocon et dégradée par le protéasome [4]. Dans ce cas, la protéine ne pouvant pas exercer son rôle physiologique, on parle de « perte de fonction ». D’autres molécules mal repliées s’accumulent dans les cellules ou dans les compartiments extracellulaires, et y forment des agrégats : c’est le cas du peptide β-amyloïde responsable de la maladie d’Alzheimer. On parle alors de « gain de fonction », ce qui peut paraître paradoxal puisqu’en général cette accumulation de protéines inactives a des conséquences cellulaires toxiques. Dans certains cas, une perte de fonction et un gain de fonction peuvent agir de concert pour provoquer un état pathologique (la rétinite pigmentaire par exemple, voir plus loin).
Le repliement défectueux de protéines qui caractérise les maladies dites « conformationnelles » [5, 6] dont il sera question dans cette revue résulte de la présence de mutations dans les gènes correspondants de ces protéines. Ces mutations sont transmissibles de génération en génération ou acquises spontanément. Quelques exemples typiques de ces maladies sont décrits dans l’Encadré 1 et le Tableau I en donne une liste plus exhaustive. Elles mettent en cause des protéines majeures de la signalisation ou du métabolisme, comme des canaux ioniques, des enzymes et des récepteurs membranaires de la famille des récepteurs couplés aux protéines G (RCPG) [7]. Ces différentes catégories de protéines sont soumises de façon très stricte au système contrôle-qualité du RE avant qu’elles ne s’engagent dans la voie de sécrétion et rejoignent leur compartiment cellulaire de fonction.
Quelques exemples de pathologies conformationnelles, protéines et mutations impliquées. *La colonne de ce tableau indique le nombre de mutations différentes responsables de la maladie considérée, ainsi que la ou les mutations les plus fréquentes. §Menkes : maladie du métabolisme du cuivre. Elle se caractérise cliniquement par un retard de croissance anté et post-natal, une détérioration neurologique progressive qui débute dans les deux premiers mois de vie, une hypotonie axiale, une spasticité, une hypothermie, des difficultés d’alimentation, des convulsions partielles ou généralisées (source : Orphanet). **Hirschsprung : malformations du tube digestif (1/5 000 naissances) résultant d’une anomalie du développement du système nerveux entérique. Elle se définit par l’absence de cellules ganglionnaires assurant l’innervation intrinsèque des couches musculaires de l’intestin terminal. Elle se traduit par une occlusion intestinale basse ou une constipation opiniâtre (source : Orphanet).
Chaperons chimiques et pharmacologiques
Extrêmement efficace dans la plupart des cas, le système contrôle-qualité du RE peut se révéler « trop » parfait. Des mutations qualifiées de mineures, ne compromettant pas la fonctionnalité des protéines impliquées, provoquent cependant soit leur déroutage et leur dégradation provoquant une maladie conformationnelle de type « perte de fonction », soit l’accumulation de la protéine cible et, dans ce cas, une pathologie conformationnelle « gain de fonction ». La compréhension du processus en cause est cruciale car l’opportunité thérapeutique existe de corriger le défaut de ciblage de la protéine mutante et de proposer un « sauvetage » ou une récupération de fonction, et donc in fine d’anticiper un traitement potentiel de la maladie en question.
Premières observations de chaperons chimiques
Le premier argument suggérant une possible « manipulation » du système contrôle-qualité du RE vient d’études effectuées sur le canal chlore CFTR (Cystic fibrosis transmembrane conductance regulator) impliqué dans la mucoviscidose. Une baisse de la température du milieu dans lequel les cellules exprimant le mutant CFTR le plus fréquent (ΔF508) croissent augmente le nombre de canaux fonctionnels à la membrane plasmique. Ce résultat fut interprété comme un effet cinétique, en l’occurrence un ralentissement du procédé de repliement de la protéine, favorisant le sauvetage du mutant ΔF508. À la suite de cette observation, les effets de petits composés chimiques, tels que le diméthylsulfoxyde (DMSO, solvant organique), le glycérol ou le triéthylamine N-oxyde (TMAO, tous deux définis comme osmolytes cellulaires), connus pour stabiliser la conformation native de plusieurs protéines et s’opposer à la dénaturation thermique ou chimique, furent testés. Les effets sur le mutant ΔF508 ont été très probants, ces composés induisant de façon très efficace la maturation de la protéine et son « échappement » au système contrôle-qualité [8]. Le mécanisme d’action de ces molécules n’est pas complètement compris : on pense qu’elles stabilisent une conformation protéique quasi native, et donc favorisent le ciblage des mutants vers le compartiment cellulaire dans lequel ils exercent leur fonction. Le glycérol, par exemple, augmente la stabilité des protéines en diminuant leur surface accessible au milieu aqueux. Ces molécules sont collectivement appelées chaperons chimiques. Aujourd’hui, l’action bénéfique de ces chaperons chimiques a été démontrée in vitro non seulement vis-à-vis du CFTR mais aussi d’autres protéines dont l’α1-antitrypsine, l’ATPase MNK (ATPase à cuivre de Menkes), l’α-galactosidase, la p53, le récepteur SUR1, l’aquaporine-2, des RCPG. Cependant, dans toutes ces études, des concentrations très élevées (en général micromolaires) de ces chaperons chimiques sont nécessaires pour entraîner un effet, et leur manque de spécificité empêche toute utilisation thérapeutique dans le cadre d’essais cliniques. Par analogie, des peptides cationiques tels que la pénétratine, qui entrent dans les cellules par un phénomène d’endocytose, possèdent la capacité de cibler vers la membrane plasmique des mutants du récepteur V2 de l’hormone anti-diurétique, responsables d’un DINc (diabète insipide néphrogénique congénital) et retenus dans des compartiments post-RE (Ergic ou endoplasmic reticulum-Golgi intermediate compartment par exemple) [9]. Dans cet exemple aussi, des concentrations micromolaires sont nécessaires et la notion de spécificité d’action vis-à-vis de la protéine cible n’est pas présente. Comme pour les chaperons chimiques, le mécanisme d’action précis n’est pas connu. Cependant, ces peptides cationiques entraînent une augmentation cytosolique de calcium et donc modulent l’activité dépendante du calcium de certains chaperons moléculaires du système contrôle-qualité cellulaire [9].
Le concept de chaperon pharmacologique
L’utilisation de chaperons chimiques a conduit naturellement les scientifiques à rechercher des molécules spécifiques utilisables dans le traitement des maladies conformationnelles chez l’homme. Une première étude fut réalisée à l’aide de mutations artificielles du gène de résistance multidrogues. Celui-ci code pour un transporteur membranaire appelé P-glycoprotéine 1 (MDR1) qui interagit avec une panoplie d’agents cytotoxiques [10]. Les mutants de MDR1 sont retenus dans le RE, mais le traitement des cellules à l’aide de substrats (vinblastine et capsaicine) ou d’inhibiteurs (cyclosporine et vérapamil) du transporteur permet une maturation du MDR1 et son ciblage à la membrane plasmique et donc une récupération de fonction. Le mécanisme proposé était le suivant : l’occupation du site actif par des composés spécifiques pourrait stabiliser un état conformationnel quasi natif accepté par le système contrôle-qualité du RE. Ce scénario implique une liaison de composés pharmacologiques (substrats, inhibiteurs) à des mutants mal repliés de protéines pour les stabiliser ; il est similaire à celui des chaperons chimiques mais fait intervenir un paramètre crucial : celui de la spécificité d’action. C’est l’acte de naissance de la notion de chaperons pharmacologiques. Selon ce concept, des ligands qui, en se fixant à des récepteurs connus, stabilisent une conformation spécifique (active, inactive), pourraient être utilisés comme chaperons pharmacologiques vis-à-vis de mutants de RCPG séquestrés dans le RE et responsables de maladies génétiques. Ainsi, l’utilisation d’antagonistes non peptidiques du récepteur V2 de la vasopressine pour neutraliser l’effet de mutations responsables du DINc, par exemple le mutant V2R Δ62-64 (délétion des résidus aminoacides 62 à 64), fut couronnée de succès [11]. Ces antagonistes (ou agonistes inverses, voir définition ci-dessous) - le SR121463, le SR49059, ou le VPA985 par exemple capables de traverser les membranes cellulaires du fait de leur nature chimique hydrophobe, ont permis une maturation des mutants testés et leur ciblage à la membrane plasmique, entraînant une récupération de fonction ici leur activation par la vasopressine et leur capacité à se coupler à la protéine Gs et à produire de l’AMPc (adénosine monophosphate, signal intracellulaire). Des antagonistes peptidiques incapables de traverser les membranes cellulaires ne peuvent pas dupliquer ces effets. La stratégie des pharmacochaperons a été appliquée depuis a de nombreuses protéines responsables de plusieurs maladies conformationnelles, mais il s’agit pour l’instant d’études cellulaires in vitro (voir Tableau II et Figure 1). Par leur spécificité d’action, les pharmacochaperons exercent leur activité à des concentrations nanomolaires [12–14], ce qui est un avantage supplémentaire par rapport aux chaperons chimiques utilisés à des concentrations cent à mille fois plus élevées. L’utilisation de chaperons pharmacologiques in vivo chez l’homme est très récente. Une étude a été réalisée chez des patients atteints de DINc et a montré que l’administration d’un antagoniste des récepteurs de la vasopressine (SR49059) permettait de di minuer fortement le volume urinaire et la prise d’eau [12]. C’est à ce jour le seul exemple publié d’application thérapeutique de chaperons pharmacologiques chez l’homme, mais il est extrêmement prometteur. La stratégie est reproduite aujourd’hui pour d’autres maladies conformationnelles, en particulier pour le traitement des maladies de Fabry et de Gaucher.
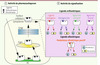 |
Figure 1 Mode d’action des chaperons pharmacologiques : cas des récepteurs membranaires. Les chaperons pharmacologiques traversent les membranes cellulaires pour interagir avec leurs récepteurs cibles mal repliés et séquestrés au niveau du RE (panneau de gauche). Le changement de conformation induit par le pharmacochaperon permet au récepteur de reprendre son trafic cellulaire classique (à travers l’Ergic et le Golgi) pour subir une maturation complète et être exporté vers son compartiment de fonction (en l’occurrence la membrane plasmique). Les chaperons pharmacologiques peuvent être de natures différentes. Différentes classes sont décrites : agonistes (activateurs des voies de signalisation, protéines G et arrestines par exemple pour les RCPG), antagonistes (ou agonistes inverses dans certains cas, inhibiteurs des voies de signalisation), agonistes biaisés (activateurs ou inhibiteurs d’une partie des voies de signalisation naturellement engagées par le ligand endogène), modulateurs allostériques positifs (permettent de potentialiser les effets du ligand endogène). |
Chaperons pharmacologiques et protéines cibles. *Des mutants naturels du récepteur δ-opioïde n’ont pas été décrits, mais le récepteur est une cible majeure dans le traitement de la douleur. Le δOR est naturellement mal maturé et 40 % uniquement des récepteurs synthétisés sont ciblés a la surface cellulaire (membrane plasmique). Il y a donc un intérêt thérapeutique réel à développer des chaperons pharmacologiques de ce récepteur [25]. **Le récepteur β1-adrénergique n’est pas impliqué dans des maladies conformationnelles, mais les chaperons pharmacologiques spécifiques sont capables de cibler des mutants artificiels de ce récepteur à la surface cellulaire [47].
Caractéristiques des chaperons pharmacologiques
Antagonistes (et agonistes inverses)
La grande majorité des chaperons pharmacologiques de RCPG décrits aujourd’hui sont des antagonistes (inhibiteurs de récepteurs) et des agonistes inverses (classés tout d’abord comme des antagonistes, ces composés sont capables de diminuer le niveau basal d’activité de leurs récepteurs spécifiques). Dans le cas de l’hypogonadisme hypogonadotrope (Encadré 1), il existe par exemple au moins quatre classes chimiques d’antagonistes pharmacochaperons du récepteur de l’hormone GnRH (gonadotrophine releasing hormone) (voir Tableau II) : indoles, quinolones, macrolides dérivés de l’érythromycine, thiéno[2,3-β]pyrimidine-2,4-diones [15]. Dans le cas de la rétinite pigmentaire, plusieurs antagonistes (agonistes inverses) pharmacochaperons ont été testés : 9-cis-rétinal, 11 -cis-rétinal et 11 -cis-7-ring rétinal [16]. Aujourd’hui, peu d’agonistes pharmacochaperons (activateurs des récepteurs) sont décrits, même si de nombreuses molécules possèdent intrinsèquement cette propriété [17]. D’un point de vue général, ces différentes catégories de chaperons pharmacologiques possèdent la capacité de traverser les membranes cellulaires pour atteindre leurs protéines cibles bloquées dans le RE ou dans un autre compartiment intracellulaire (Figure 1). Ces molécules sont donc plutôt de nature lipophile, et leur activité de pharmacochaperon ne peut pas être reproduite par des ligands non perméants. La restauration de la fonction d’une protéine mutante dépend bien sûr de la mutation elle-même et de ses conséquences sur la conformation globale. Mais de façon intéressante, il existe une corrélation entre l’affinité (force d’interaction) du chaperon pharmacologique pour le récepteur et la récupération de la fonction protéique. Plus l’affinité est forte, plus l’effet est important. Intuitivement, cette corrélation ne va pas de soi pour les antagonistes et les agonistes inverses. En effet, que le chaperon agisse comme antagoniste ou agoniste inverse, la récupération de la fonction protéique est la résultante d’un équilibre subtil entre la force du caractère chaperon et la possibilité qu’il soit déplacé par le ligand endogène naturel activateur. C’est tout le paradoxe de ces ligands qui permettent un ciblage des récepteurs mutants à la surface cellulaire mais qui sont dans l’impossibilité de stimuler directement la protéine dont ils sont inhibiteurs. Pour une application thérapeutique, il semblerait donc plus approprié de sélectionner des chaperons pharmacologiques antagonistes et agonistes inverses possédant une affinité modérée pour leur cible. Un exemple qui illustre bien ce concept est celui de l’antagoniste SR49059 non spécifique du récepteur V2 (mais spécifique du récepteur V1a de la vasopressine) et pour lequel il a une affinité très moyenne. Cependant, ce ligand est capable de cibler des mutants du V2R à la surface cellulaire et d’induire un effet antidiurétique chez des malades atteints de DINc [12].
Agonistes
Logiquement, les chaperons pharmacologiques agonistes seraient donc plus intéressants puisque capables à la fois de cibler les récepteurs mal repliés à la surface et de les stimuler directement (Figure 1). Dans ce cas, une forte affinité est un avantage, et ces molécules peuvent remplacer avantageusement le ligand naturel activateur. Mais les agonistes présentent aussi des inconvénients. Leur liaison avec le récepteur cible provoque son activation mais également son internalisation via une interaction avec une protéine régulatrice, l’arrestine. L’internalisation, qui fait intervenir un mécanisme d’endocytose, régule le nombre de récepteurs à la surface cellulaire et donc l’intensité du signal intracellulaire. Une fois le récepteur internalisé, il n’est plus accessible au ligand activateur. Ce phénomène (appelé aussi processus de désensibilisation) conduit à l’arrêt progressif de la réponse cellulaire. Des agonistes du récepteur δ-opioïde (δOR) et d’un mutant artificiel de cette protéine (D95A), possédant une activité de pharmacochaperon, ont été décrits mais ils ont également la capacité d’induire l’internalisation du δOR via l’arrestine [17].
Agonistes biaisés
La découverte très récente d’agonistes biaisés du V2R (MCF14, MCF18, MCF57) offre une nouvelle opportunité thérapeutique dans le DINc [13]. Un agoniste biaisé est une molécule qui ne possède qu’une partie des propriétés pharmacologiques du ligand activateur endogène. En l’occurrence, les chaperons pharmacologiques MCF14, MCF18 et MCF57 de plusieurs mutants du V2R sont des activateurs du récepteur : ils provoquent l’activation de la protéine Gs, mais n’entraînent pas son internalisation parce qu’ils s’opposent au recrutement de l’arrestine (Figure 1). Ils offrent donc un avantage certain par rapport aux agonistes classiques et aux antagonistes, tout en ayant une affinité forte vis-à-vis du V2R. L’association de ces caractéristiques semble idéale pour traiter les patients atteints de DINc puisque ces molécules induisent effectivement le ciblage de mutants du V2R à la surface cellulaire [13]. Ces composés devraient permettre a priori une activation soutenue et durable dans le temps des V2R mutés puisque ceux-ci ne sont pas internalisés. On peut imaginer, même si cela reste à démontrer, un traitement par voie orale peu contraignant pour les malades, et espérer une régulation efficace de la diurèse.
Modulateurs allostériques
Enfin, il nous faut citer une dernière catégorie de molécules, les modulateurs allostériques, qui pourraient également être des chaperons pharmacologiques tout aussi prometteurs. Un modulateur allostérique se fixe sur sa protéine cible de façon spécifique, mais dans un site d’interaction différent de celui du substrat naturel (pour les enzymes) ou du ligand endogène (pour les récepteurs ou les canaux ioniques). Sa liaison à la protéine entraîne un changement de conformation qui peut avoir une influence positive ou négative sur la liaison de l’activateur (ligand, substrat), sur la capacité de la protéine à produire un effet biologique, ou sur les deux (Figure 1). Il n’y a qu’un exemple aujourd’hui de modulateur allostérique pouvant agir comme chaperon pharmacologique. Cette molécule est spécifique du récepteur de l’hormone FSH (follicle stimulating hormone) [18]. Lorsqu’elle se fixe sur son récepteur, elle participe à la régulation de la maturation des follicules ovariens chez la femme et contrôle la spermatogenèse chez l’homme. Plusieurs mutations du récepteur de la FSH ont été découvertes qui provoquent des troubles endocriniens. La mutation A189V bloque le récepteur dans le RE et empêche son ciblage à la membrane plasmique. Un composé activateur spécifique du récepteur a été découvert, mais il n’est pas un ligand compétiteur de l’hormone naturelle. Ce composé (Org41841, famille des thiénopyr(im)idines) permet l’acheminement du récepteur mutant à la surface cellulaire où il peut être activé et induire une réponse de la cellule. Dans ce cas, Org41841 et FSH agissent de concert : Org41841 favorise l’activité de l’hormone FSH. Ce type de molécule possède donc des caractéristiques tout à fait particulières, c’est un modulateur allostérique positif avec une activité de chaperon pharmacologique.
Perspectives
Le concept de chaperon pharmacologique découle directement de l’idée que toute mutation dans une protéine n’entraîne pas obligatoirement une perte de fonction, à condition que le défaut mineur puisse être sélectivement compensé. En effet, beaucoup de protéines mutées pourraient être actives mais sont tout simplement rejetées par le système contrôle-qualité du RE et ne sont pas exportées dans leur compartiment cellulaire final. Or, un nombre croissant d’exemples démontrent que les chaperons pharmacologiques facilitent le repliement et la maturation de leurs protéines cibles, in vitro comme in vivo, y compris chez des patients. Il est donc probable que cette stratégie thérapeutique puisse être applicable à une majorité de maladies conformationnelles. Ces outils thérapeutiques prometteurs concernent des pathologies conformationnelles aussi bien « perte de fonction » que « gain de fonction » en prévenant l’agrégation des protéines mal repliées. De plus, le fait qu’un chaperon pharmacologique permette une récupération de fonction de plusieurs mutants d’une même protéine spécifique devrait simplifier le développement de médicaments ou drogues utiles pour traiter les maladies conformationnelles. En d’autres termes, il n’est pas nécessaire de développer un médicament chaperon pharmacologique pour chaque mutation (chaque patient). Ceci est un élément important qui devrait inciter les industriels à s’investir plus activement dans la mise au point de ces molécules. De façon intéressante, les chaperons pharmacologiques agissent sur des protéines dont beaucoup sont des cibles thérapeutiques majeures (récepteurs, canaux, transporteurs, enzymes). De multiples ligands avec une forte affinité et sélectivité sont déjà identifiés ou en développement. Ces ligands sont des pharmacochaperons en devenir, à la condition qu’ils soient capables de traverser les membranes cellulaires pour atteindre leurs cibles bloquées dans le RE ou dans d’autres compartiments (Ergic, Golgi). Les futures campagnes de criblage de chaperons pharmacologiques devront donc tirer parti de chimiothèques déjà disponibles et dont les cibles sont bien définies. La recherche d’agonistes biaisés et de modulateurs allostériques positifs sera particulièrement attractive. Le fait que de très nombreuses molécules soient déjà en développement, même si leur caractère pharmacochaperon n’est pas pour l’instant la propriété thérapeutique mise en avant, est vraiment un avantage. Ces médicaments à caractère pharmacochaperon potentiel seront développés en priorité pour des pathologies majeures bien définies. Mais ils pourraient aussi être testés en parallèle sur des patients volontaires atteints de maladies orphelines, lorsque leur processus de développement clinique sera suffisamment avancé (phase III), et ce malgré la réticence des entreprises pharmaceutiques à mettre sur le marché des médicaments pour les maladies orphelines.
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Les auteurs remercient Muriel Asari-Gien pour la réalisation iconographique de cet article.
Références
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nature Rev Mol Cell Biol 2003 ; 4 : 181-91. [Google Scholar]
- Anelli T, Sitia R. Protein quality control in the early secretory pathway. EMBOJ 2008 ; 27 : 315-27. [Google Scholar]
- Arrigo AP. Chaperons moléculaires et repliement des protéines. MedSci (Paris) 2005 ; 21 : 619-25. [Google Scholar]
- Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nature Rev Mol Cell Biol 2002 ; 3 : 246-55. [Google Scholar]
- Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolfding diseases. Nature 2003 ; 426 : 905-9. [Google Scholar]
- Chaudhuri TK, Paul S. Protein-misfolding diseases and chaperone-based therapeutic approaches. FEBSJ 2006 ; 273 : 1331-49. [Google Scholar]
- Conn PM, Ulloa-Aguirre A, Ito J, Janovick JA. G protein-coupled receptors trafficking in health and disease : lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol Rev 2007 ; 59 : 225-50. [Google Scholar]
- Sato S, Ward CL, Krouse ME, et al. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem 1996 ; 271 : 635-8. [Google Scholar]
- Oueslati M, Hermosilla R, Schönenberger E, et al. Rescue of a nephrogenic diabetes insipiduscausing vasopressin V2 receptor mutant by cell-penetrating peptides. J Biol Chem 2007 ; 282 : 20676-85. [Google Scholar]
- Loo TW, Clarke DM. Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J Biol Chem 1997 ; 272 : 709-12. [Google Scholar]
- Morello JP, Salahpour A, Laperrière A, et al. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest 2000 ; 105 : 887-95. [Google Scholar]
- Bernier V, Morello JP, Zarruk A, et al. Pharmacologic chaperones as a potential treatment for Xlinked nephrogenic diabetes insipidus. J Am Soc Nephrol 2006 ; 17 : 232-43. [Google Scholar]
- Jean-Alphonse F, Perkovska S, Frantz MC, et al. Biased agonist pharmacochaperones of the AVP V2 receptor may treat congenital nephrogenic diabetes insipidus. J Am Soc Nephrol 2009 ; 20 : 2190-203. [Google Scholar]
- Robben JH, Sze M, Knoers NV, Deen PM. Functional rescue of vasopressin V2 receptor mutants in MDCK cells by pharmacochaperones: relevance to therapy of nephrogenic diabetes insipidus. Am J Physiol Renal Physiol 2007 ; 292 : F253-60. [Google Scholar]
- Conn PM, Janovick JA. Drug development and the cellular quality control system. Trends Pharmacol Sci 2009 ; 30 : 228-33. [Google Scholar]
- Li T, Sandberg MA, Pawlyk BS, et al. Effect of vitamine A supplementation on rhodopsin mutants T17M and P347S in transgenic mice and in cell cultures. Proc Natl Acad Sci USA 1998 ; 95 : 11933-8. [Google Scholar]
- Petäjä-Repo UE, Hogue M, Bhalla S, et al. Ligands act as pharmacological chaperones and increase the efficiency of δ opioid receptor maturation. EMBO J 2002 ; 21 : 1628-37. [Google Scholar]
- Janovick JA, Maya-Nunez G, Ulloa-Aguirre A, et al. Increased plasma membrane expression of human follicle-stimulating hormone receptor by a small molecule thienopyr(im)idine. Mol Cell Endocrinol 2009 ; 298 : 84-8. [Google Scholar]
- Riordan JR. The cystic fibrosis transmembrane conductance regulator. Annu Rev Physiol 1993 ; 55 : 609-30. [Google Scholar]
- Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene : genetic analysis. Science 1989 ; 245 : 1073-80. [Google Scholar]
- Germain DP. La maladie de Fabry : de la découverte des lysosomes à l’avènement de la thérapeutique. Med Sci (Paris) 2005 ; 21 : 5-7. [Google Scholar]
- Fan JQ, Ishii S, Asano N, Suzuki Y. Accelerated transport and maturation of lysosomal alphagalactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med 1999 ; 5 : 112-5. [Google Scholar]
- Grabowski GA. Gaucher disease: enzymology, genetics and treatment. Adv Hum Genet 1993 ; 21 : 377-441. [Google Scholar]
- Sawkar AR, Cheng WC, Beutler E, et al. Chemical chaperones increase the cellular activity of N370S beta-glucosidase: a therapeutic strategy for Gaucher disease. Proc Natl Acad Sci USA 2002 ; 99 : 15428-33. [Google Scholar]
- Mendes HF, Van der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa : implications for therapy. Trends Mol Med 2005 ; 11 : 177-85. [Google Scholar]
- Morello JP, Bichet DG. Nephrogenic diabetes insipidus. Annu Rev Physiol 2001 ; 63 : 607-30. [Google Scholar]
- Carrell RW, Lomas DA, Sidhar S, Foreman R. alpha1-antitrypsin deficiency: a conformational disease. Chest 1996 ; 110 : 243S-47S. [Google Scholar]
- Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science 1999 ; 286 : 2507-10. [Google Scholar]
- Toledo F, Bluteau O, Simeonova I. Réactivation de p53 dans les tumeurs : une stratégie antitumorale prometteuse. Med Sci (Paris) 2007 ; 23 : 565-7. [Google Scholar]
- Lam CW, Xie J, To KF, et al. A frequent activated smoothened mutation in sporadic basal cell carcinomas. Oncogene 1999 ; 18 : 833-6. [Google Scholar]
- Partridge CJ, Beech DJ, Sivaprasadarao A. Identification and pharmacological correction of a membrane trafficking defect associated with a mutation in the sulfonylurea receptor causing familial hyperinsulinism. J Biol Chem 2001 ; 276 : 35947-52. [Google Scholar]
- Pratt EB, Yan FF, Gay JW, et al. Sulfonylurea receptor 1 mutations that cause opposite insulin secretion defects with chemical chaperone exposure. J Biol Chem 2009 ; 284 : 7951-9. [Google Scholar]
- Kim BE, Smith K, Meagher CK, Petrisd MJ. A conditional mutation affecting localization of the Menkes disease copper ATPase ; suppression by copper supplementation. J Biol Chem 2002 ; 277 : 44079-84. [Google Scholar]
- Liu X, Garriga P, Khorana HG. Structure and function in rhodopsin : correct folding and misfolding in two point mutants in the intradiscal domain of rhodopsin identified in retinitis pigmentosa. Proc Natl Acad Sci USA 1996 ; 93 : 4554-9. [Google Scholar]
- Fuchs S, Amiel J, Claudel S, et al. Functional characterization of three mutations of the endothelin B receptor gene in patients with Hirshsprung’s disease: evidence for selective loss of Gi coupling. Mol Med 2001 ; 7 : 115-24. [Google Scholar]
- Lubrano-Berthelier C, Dubern B, Lacorte JM, et al. Melanocortin 4 receptor mutations in a large cohort of severely obese adults : : prevalence, functional classification, genotype-phenotype relationship, and lack of association with binge eating. J Clin EndocrinolMetab 2006 ; 91 : 1811-8. [Google Scholar]
- Clark AJL, Metherell LA, Cheetham ME, Huebner A. Inherited ACTH insensitivity illuminates the mechanisms of ACTH action. Trends Endocrinol Metab 2005 ; 16 : 451-7. [Google Scholar]
- Samson M, Libert F, Doranz BJ, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996 ; 382 : 722-5. [Google Scholar]
- Tfelt-Hansen J, Brown EM. The clacium-sensing receptor in normal physiology and athophysiology: a review. Crit Rev Clin Lab Sci 2007 ; 42 : 35-70. [Google Scholar]
- Biebermann H, Schoneberg T, Krude H, et al. Mutations of the human thyrotropin receptor gene causing thyroid hypoplasia and persistent congenital hypothyroidism. J Clin Endocrinol Metab 1997 ; 82 : 3471-80. [Google Scholar]
- Themmen APN, Martens JWM, Brunner HG. Activating and inactivating mutations in LH receptors. Mol Cell Endocrinol 1998 ; 145 : 137-42. [Google Scholar]
- Aittomäki KJ, Lucena LD, Pakarinen P, et al. Mutation in the folliclestimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell 1995 ; 82 : 959-68. [Google Scholar]
- Beaumont KA, Newton RA, Smit DJ, et al. Altered cell surface expression of human MCR1 variant receptor alleles associated with red hair and skin cancer risk. Hum Mol Genet 2005 ; 14 : : 2145-54. [Google Scholar]
- Noorwez SM, Malhotra R, McDowell JH, et al. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J Biol Chem 2004 ; 279 : 16278-84. [Google Scholar]
- Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet 2009 ; 373 : 2055-66. [Google Scholar]
- Sacchetini JC, Kelly JW. Therapeutic strategies for human amyloid diseases. Nat Rev Drug Discov 2002 ; 1 : 267-75. [Google Scholar]
- Kobayashi H, Ogawa K, Yao R, et al. Functional rescue of beta-adrenoceptor dimerization and trafficking by pharmacological chaperones. Traffic 2009 ; 10 : 1019-33. [Google Scholar]
- Stanton BZ, Peng LF. Small molecule modulators of the Sonic Hedgehog signalling pathway. Mol Biosyst 2010 ; 6 : 44-54. [Google Scholar]
- Pey AL, Ying M, Cremades N, et al. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J Clin Invest 2008 ; 118 : 2858-67. [Google Scholar]
- Asano N, Ishii S, Kizu H, et al. In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin an dits derivatives. Eur J Biochem 2000 ; 267 : 4179-86. [Google Scholar]
- Loo TW, Bartlett MC, Wang Y, Clarke DM. Rescue of deltaF508 and other misprocessed CFTR mutants by a novel quinazoline compound. Mol Pharmacol 2005 ; 2 : 407-13. [Google Scholar]
- Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA 2001 ; 98 : 9836-41. [Google Scholar]
- Sigurdsson EM, Permanne B, Soto C, et al. In vivo reversal of amyloid-beta lesions in rat brain. J Neuropathol Exp Neurol 2000 ; 59 : 11-7. [Google Scholar]
- Heiser V, Scherzinger E, Boeddrich A, et al. Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: implications for huntington’s disease therapy. Proc Natl Acad Sci USA 2000 ; 97 : 6739-44. [Google Scholar]
Liste des tableaux
Quelques exemples de pathologies conformationnelles, protéines et mutations impliquées. *La colonne de ce tableau indique le nombre de mutations différentes responsables de la maladie considérée, ainsi que la ou les mutations les plus fréquentes. §Menkes : maladie du métabolisme du cuivre. Elle se caractérise cliniquement par un retard de croissance anté et post-natal, une détérioration neurologique progressive qui débute dans les deux premiers mois de vie, une hypotonie axiale, une spasticité, une hypothermie, des difficultés d’alimentation, des convulsions partielles ou généralisées (source : Orphanet). **Hirschsprung : malformations du tube digestif (1/5 000 naissances) résultant d’une anomalie du développement du système nerveux entérique. Elle se définit par l’absence de cellules ganglionnaires assurant l’innervation intrinsèque des couches musculaires de l’intestin terminal. Elle se traduit par une occlusion intestinale basse ou une constipation opiniâtre (source : Orphanet).
Chaperons pharmacologiques et protéines cibles. *Des mutants naturels du récepteur δ-opioïde n’ont pas été décrits, mais le récepteur est une cible majeure dans le traitement de la douleur. Le δOR est naturellement mal maturé et 40 % uniquement des récepteurs synthétisés sont ciblés a la surface cellulaire (membrane plasmique). Il y a donc un intérêt thérapeutique réel à développer des chaperons pharmacologiques de ce récepteur [25]. **Le récepteur β1-adrénergique n’est pas impliqué dans des maladies conformationnelles, mais les chaperons pharmacologiques spécifiques sont capables de cibler des mutants artificiels de ce récepteur à la surface cellulaire [47].
Liste des figures
|
Figure 1 Mode d’action des chaperons pharmacologiques : cas des récepteurs membranaires. Les chaperons pharmacologiques traversent les membranes cellulaires pour interagir avec leurs récepteurs cibles mal repliés et séquestrés au niveau du RE (panneau de gauche). Le changement de conformation induit par le pharmacochaperon permet au récepteur de reprendre son trafic cellulaire classique (à travers l’Ergic et le Golgi) pour subir une maturation complète et être exporté vers son compartiment de fonction (en l’occurrence la membrane plasmique). Les chaperons pharmacologiques peuvent être de natures différentes. Différentes classes sont décrites : agonistes (activateurs des voies de signalisation, protéines G et arrestines par exemple pour les RCPG), antagonistes (ou agonistes inverses dans certains cas, inhibiteurs des voies de signalisation), agonistes biaisés (activateurs ou inhibiteurs d’une partie des voies de signalisation naturellement engagées par le ligand endogène), modulateurs allostériques positifs (permettent de potentialiser les effets du ligand endogène). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.