")
")
| Issue |
Med Sci (Paris)
Volume 26, Number 5, Mai 2010
|
|
|---|---|---|
| Page(s) | 516 - 521 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/2010265516 | |
| Published online | 15 mai 2010 | |
GSK-3β : une kinase au cœur des maladies neuro-dégénératives ?
GSK-3β: a central kinase for neurodegenerative diseases ?
Institut de pharmacologie moléculaire et cellulaire, 660, route des Lucioles, Sophia Antipolis, 06560 Valbonne, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Le vieillissement de la population entraîne une augmentation spectaculaire de l’incidence des maladies neurodégénératives dans les pays industrialisés. L’implication de la GSK-3β (glycogen synthase kinase) dans la pathogenèse de la maladie d’Alzheimer est bien établie, mais il semble également évident que la dérégulation de l’activité de la GSK-3β pourrait être étroitement liée à d’autres maladies neurodégénératives, comme les maladies de Parkinson et de Huntington. Dans cette revue, je résumerai les découvertes permettant de définir l’activité de la GSK-3β comme étant le point de convergence des processus à l’origine de ces maladies. L’inhibition de l’activité de la GSK-3β en tant qu’approche thérapeutique possible sera également discutée.
Abstract
Neurodegenerative diseases are more and more prevalent in our aging societies. There is strong evidence that glycogen synthase kinase (GSK)-3β plays a crucial role in Alzheimer’s disease (AD). Indeed, it is involved in the regulation of the two major neuropathological hallmarks present in the brains of AD patients. Interestingly, the kinase has been implicated in multiple cellular processes and linked with the pathogenesis and neuronal loss in several neurodegenerative diseases, including Parkinson’s and Huntington’s diseases, in which abnormally elevated levels of GSK-3β activity have been reported. In this review, we will provide an overview of the current data pointing out the convergent role of GSK-3β in the neuropathological pathways of these diseases. We will also discuss the rationale for the development of specific inhibitors with therapeutic potentials for such devastating human diseases.
© 2010 médecine/sciences - Inserm / SRMS
La GSK (glycogen synthase kinase)-3 est une sérine/thréonine kinase initialement découverte pour son rôle majeur dans la signalisation intracellulaire induite par l’insuline. Chez les mammifères, il existe deux isoformes très proches : la GSK-3α et la GSK-3β. Récemment, la GSK-3β s’est imposée comme étant une cible thérapeutique prometteuse dans les maladies neurodégénératives. La GSK-3β, très exprimée dans les neurones, a la particularité d’être constitutivement active ; elle est négativement régulée par phosphorylation sur son résidu sérine 9. Son activité est inhibée par plusieurs stimulus extérieurs, dont l’insuline, les ligands Wnt et la voie Akt. La GSK-3β est également phosphorylée sur la tyrosine 216, probablement par autophosphorylation, ce qui augmente son activité [1]. Parmi les nombreux substrats de la kinase figure un grand nombre de protéines essentielles au développement des maladies neurodégénératives, dont les protéines Tau, l’α-synucléine, la synphiline-1 et la protéine précurseur de l’amyloïde. Cette revue se focalisera sur le rôle central que la GSK-3β joue dans les maladies neurodégénératives majeures (Figure 1).
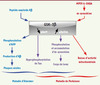 |
Figure 1. Représentation schématique du rôle central de la GSK-3β dans les voies neurodégénératives aboutissant à la mort neuronale. Diverses neurotoxines, dont le peptide amyloïde Aβ impliqué dans la maladie d’Alzheimer, le MPTP et la 6-hydroxydopamine (6-OHDA) dont les effets miment la maladie de Parkinson, activent, directement ou indirectement, l’activité de la GSK-3β en augmentant la phosphorylation du résidu tyrosine 216 (pTyr216) et en inhibant la phosphorylation du résidu sérine 9 (pSer9). L’activation de la GSK-3 β par le peptide amyloïde a pour conséquence l’hyperphosphorylation de la protéine Tau à l’origine des dégénérescences neurofibrillaires dans la maladie d’Alzheimer ainsi que l’augmentation de la production du peptide amyloïde lui-même, ce qui aboutit à la formation de plaques séniles. Les deux lésions histopathologiques caractéristiques de la maladie d’Alzheimer sont les agrégats de protéine Tau hyperphosphorylée et les plaques séniles. L’activation de la GSK-3β dépendante de l’α-synucléine par le MPTP et la 6-OHDA entraîne l’hyperphosphorylation de la protéine Tau et son agrégation (détectée dans le cerveau de nombreux patients parkinsoniens), la phosphorylation et l’agrégation de l’α-synucléine au sein des corps de Lewy et la baisse de l’activité mitochondriale dans les neurones touchés. Tous ces phénomènes aboutissent à la mort neuronale dans des modèles cellulaires et animaux de la maladie de Parkinson. |
GSK-3β et maladie d’Alzheimer (MA)
Les trois principales caractéristiques histopathologiques de la maladie sont les plaques amyloïdes, les dégénérescences neurofibrillaires (DNF) et la mort des neurones cholinergiques. À ce jour, les liens entre ces différentes lésions sont encore mal connus et débattus, ce qui rend le traitement de la MA difficile. De manière intéressante, la GSK-3β est impliquée dans au moins deux de ces processus pathologiques (Figure 2).
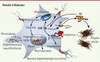 |
Figure 2. Représentation schématique de l’implication de la GSK-3β dans les voies cellulaires mises en jeu dans la maladie d’Alzheimer. Au cours de la maladie d’Alzheimer, la GSK-3β influence le clivage de la protéine précurseur de l’amyloïde (APP) par la γ-sécrétase et favorise ainsi la production du peptide amyloïde Aβ qui s’accumule dans l’espace extracellulaire et s’agrège sous la forme de plaques séniles. En retour, l’Aβ active la GSK-3β, entraînant l’hyperphosphorylation de la protéine Tau qui se dissocie des microtubules et provoque l’apparition de dégénérescences neurofibrillaires. La GSK-3β interagit également avec un membre du complexe γ-sécrétase, la préséniline-1, qui elle-même interagit avec la β-caténine. La β-caténine est phosphorylée par la GSK-3β, ce qui induit sa dégradation par le protéasome. Enfin, la GSK-3β activée favorise la mort neuronale en activant la voie apoptotique impliquant le relargage du cytochrome c, l’activation de la cascade des caspases et la fragmentation de l’ADN. |
Dégénérescences neurofibrillaires
Les DNF sont des lésions intracellulaires résultant de modifications de la protéine Tau, protéine structurale qui participe à la stabilisation du cytosquelette neuronal [41]. Au cours de la MA, la protéine Tau subit une hyperphosphorylation, se détache alors des microtubules et forme des paires de filaments hélicoïdaux pathologiques aboutissant à des DNF progressives. La GSK-3β est une des principales kinases phosphorylant Tau in vivo. L’augmentation de l’activité de la GSK-3β dans les cerveaux de patients atteints de MA coïncide au niveau spatiotemporel avec les DNF [2]. La GSK-3β surexprimée dans le cerveau de souris transgéniques provoque l’hyperphosphorylation de la protéine Tau, le désassemblage des microtubules et l’apparition de DNF [3, 4].
Plaques séniles et peptide amyloïde
La GSK-3β est impliquée dans la régulation du clivage protéolytique de l’APP (précurseur de la protéine amyloïde) et la production du peptide amyloïde (Aβ), un petit peptide toxique issu de coupures successives de l’APP par les sécrétases et principal constituant des plaques séniles. In vitro, la GSK-3β phosphoryle l’APP [5]. In vivo, dans des cerveaux de souris transgéniques surexprimant l’APP, l’inhibition de la GSK-3β par des inhibiteurs plus ou moins spécifiques, comme le lithium ou le valproate, diminue la production d’Aβ et protège les neurones contre la toxicité du peptide [6, 7]. L’Aβ lui-même aurait la capacité d’activer la GSK-3β [8]. Il favoriserait ainsi l’hyperphosphorylation de la protéine Tau, fournissant un lien mécanistique entre les deux grands stigmates de la MA que sont l’accumulation d’Aβ et les DNF.
Interaction entre les présénilines et GSK-3β
Les mutations des gènes des présénilines 1 et 2 (PS-1 et PS-2) sont responsables de formes familiales précoces de MA (environ 10 % des cas de MA). Les présénilines participent à l’activité de la γ-sécrétase, complexe multiprotéique responsable de la libération de l’Aβ à partir d’APP [9]. PS-1 interagit à la fois avec la GSK-3β et la protéine Tau. La capacité d’interaction entre GSK-3β et PS-1 est accrue lorsque cette dernière est mutée. La PS-1 permettrait donc de rapprocher la GSK-3β de l’un de ses substrats, la protéine Tau, dont la phosphorylation serait ainsi facilitée [10]. De plus, PS-1 et la β-caténine1 interagissent sous forme de complexes, stabilisant ainsi le taux de forme libre de β-caténine. Les mutations de PS-1 abolissent cet effet, favorisent la phosphorylation de la β-caténine par la GSK-3β et bloquent ainsi la voie de signalisation dépendante de Wnt, provoquant alors la mort cellulaire [11]. Ainsi, par le biais des présénilines, la GSK-3β semble impliquée dans la production d’Aβ, la régulation de la phosphorylation de Tau et la modulation des niveaux de β-caténine (Figure 2).
Potentiel thérapeutique des inhibiteurs de GSK-3β
Le lithium, couramment utilisé dans le traitement des désordres bipolaires et de la dépression, est un inhibiteur de la GSK-3β et son action a été testée dans de nombreuses études. Des doses cliniquement tolérables de lithium protègent les neurones de la mort induite par l’Aβ et réduisent sa production dans les cerveaux de souris transgéniques surexprimant des formes mutées d’APP [7, 12]. De plus, le lithium réduit l’agrégation de la protéine Tau et la mort neuronale dans des modèles de souris transgéniques de tauopathies (maladies dégénératives liées à l’agrégation de Tau), non seulement dès l’apparition des premiers signes de la neuropathologie mais aussi aux stades plus avancés de la maladie [13, 14]. Le valproate, autre inhibiteur de GSK-3β utilisé dans le traitement des désordres bipolaires et comme anticonvulsivant, a prouvé son efficacité dans un modèle de souris double-transgéniques surexprimant une forme mutée d’APP et de Tau. Le valproate diminue la quantité d’Aβ, réduit la formation des plaques séniles et diminue les déficits mnésiques de ces animaux [6].
Toutefois, le potentiel thérapeutique des inhibiteurs de GSK-3β a été remis en cause par deux études récentes. Bien que l’inhibition de la kinase soit efficace vis-à-vis des perturbations cellulaires caractéristiques de tauopathie, elle n’améliore pas les déficits de mémoire de souris modèles de MA atteintes d’une neuropathologie avancée, ce qui indique que le traitement doit débuter dans les stades précoces de la maladie [15]. De même, dans un modèle murin où la pathologie est induite par l’infusion d’oligomères d’A, un inhibiteur de GSK-3β permet de corriger certains déficits comportementaux, mais ne permet pas de récupérer les facultés de mémorisation [16].
De façon consensuelle, il est admis que l’activité aberrante de la GSK-3β promeut la mort neuronale et inhibe la prolifération cellulaire [17]. En revanche, l’activité constitutive de la GSK-3β serait essentielle à la viabilité neuronale [18] et à la potentialisation à long terme dans l’hippocampe, phénomène impliqué dans les processus de mémorisation [19]. Ainsi, le traitement d’individus sains avec des inhibiteurs de GSK-3β altèrerait leurs performances lors d’exercices de tâches verbales et de mémorisation [20].
Le lithium et le valproate n’ont pas pour unique cible la GSK-3β. C’est la raison pour laquelle d’autres composés plus spécifiques font l’objet d’intenses recherches. Les thiadiazolidinediones hétérocycliques, dont fait partie le NP-12, sont de prometteurs inhibiteurs sélectifs de GSK-3β. Ils réduisent l’hyperphosphorylation de la protéine Tau, le nombre de plaques séniles, la mort neuronale ainsi que la prolifération astrogliale et les déficits cognitifs de souris transgéniques [21]. En 2008 a démarré la phase II de l’étude clinique du NP-12 dans le traitement de la MA.
GSK-3β et maladie de Parkinson
La maladie de Parkinson (MP) se traduit, au niveau cérébral, par la mort des neurones dopaminergiques de la substance noire et l’apparition, dans les neurones survivants, de lésions appelées corps de Lewy2, composées notamment d’α-synucléine3 et de synphiline-14 [42]. Un dysfonctionnement mitochondrial serait à l’origine de la mort des neurones dopaminergiques [22]. De manière intéressante, l’activité de la GSK-3β est augmentée dans le striatum de patients parkinsoniens [23].
Les modèles de souris traitées par les neurotoxines
Le métabolite actif du MPTP (1-méthyl-1,2,3,6-tétrahydropyridine), le MPP+, est un inhibiteur du complexe I de la chaîne respiratoire mitochondriale. Ce composé est couramment utilisé pour provoquer la mort neuronale dans des modèles cellulaires et animaux [43]. L’inhibition de la GSK-3β protège les neurones dopaminergiques de la toxicité du MPTP, suggérant un rôle majeur de la kinase dans la pathogenèse de la maladie [24]. Deux autres neurotoxines dont les effets miment la MP, la 6-hydroxydopamine et la roténone, induisent également une mort neuronale dépendante de la GSK-3β. Notre équipe a montré que le MPTP/MPP+ induit l’activation de la GSK-3β aussi bien in vivo que dans des cultures primaires de neurones [25].
Interaction entre α-synucléine et GSK-3β
L’α-synucléine est le principal composant des corps de Lewy [42]. Un lien étroit semble exister entre l’α-synucléine et la protéine Tau. Ainsi, des inclusions d’α-synucléine de type corps de Lewy ont été trouvées dans le cerveau de patients atteints de MA. Inversement, des agrégats de protéine Tau sont fréquemment détectés à la périphérie des corps de Lewy présents dans des cerveaux atteints de MP ou de démence à corps de Lewy [26]. Dans le cerveau de patients atteints d’une forme génétique de MP due à une mutation de l’α-synucléine [42], des agrégats composés de Tau et de synucléine ont été trouvés [27]. In vitro et in vivo, la GSK-3β est activée par le MPTP de manière strictement dépendante de la présence de l’α-synucléine, et est alors responsable de l’hyperphosphorylation de la protéine Tau (Figure 3). L’inhibition de la GSK-3β permet de diminuer à la fois l’hyperphosphorylation de Tau et l’accumulation de l’α-synucléine [28].
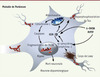 |
Figure 3. Représentation schématique de l’implication de la GSK-3β dans les voies cellulaires impliquées dans la maladie de Parkinson. Dans les modèles de maladie de Parkinson par intoxication à la neurotoxine MPTP, la GSK-3β est activée par l’intermédiaire de l’α-synucléine. En retour, la GSK-3β favorise l’agrégation de l’α-synucléine et de la synphiline-1 au sein des corps de Lewy. La 6-hydroxydopamine (6-OHDA) induit l’activation, directe ou indirecte, de la GSK-3β. La GSK-3β activée est responsable de l’hyperphosphorylation de la protéine Tau. Enfin, sous l’effet des neurotoxines, la GSK-3β activée provoque la chute du potentiel membranaire mitochondrial (Ψm), entraînant l’ouverture du pore de transition de perméabilité mitochondrial, ce qui aboutit au relargage du cytochrome c dans le cytosol, à l’activation de la cascade des caspases, à la fragmentation de l’ADN et à la mort neuronale par apoptose. |
GSK-3β phosphoryle la synphiline-1
La synphiline-1 interagit, in vivo, avec l’α-synucléine [29]. On la retrouve également au cœur des corps de Lewy. La synphiline-1 soluble serait toxique et inhiberait l’activité du protéasome (complexe enzymatique chargé de la dégradation sélective de certaines protéines, pour revue [30]). En revanche, elle serait protectrice sous sa forme agrégée puisque des cellules présentant les inclusions ne s’engagent pas dans la voie apoptotique. In vitro et in vivo, la synphiline-1 est phosphorylée par la GSK-3b, entraînant une diminution de sa dégradation par le protéasome. L’absence de phosphorylation de la synphiline-1 diminuerait la quantité de sa forme soluble, favorisant la formation des inclusions [31]. Ceci indique que la GSK-3β jouerait un rôle dans l’agrégation de la synphiline-1 dans les corps de Lewy (Figure 3).
GSK-3β et métabolisme des mitochondries
L’implication des mitochondries dans la pathogenèse de la MP est bien établie. La GSK-3β est présente dans le cytosol, le noyau, mais aussi dans les mitochondries, où son rôle demeure méconnu. Notre étude montre que la GSK-3β agirait sur l’induction de la voie apoptotique mitochondriale déclenchée par le MPTP. En effet, le MPP+ provoque l’activation de la GSK-3β cytosolique et l’inhibition de la GSK-3β mitochondriale. Ce phénomène provoque la chute du potentiel membranaire de la mitochondrie et l’activation de la voie apoptotique mitochondriale aboutissant à la mort neuronale (Figure 3). L’inhibition de la GSK-3β par deux inhibiteurs différents ou le traitement par des siARN abolit tous ces effets néfastes [25]. La GSK-3β serait également impliquée dans l’ouverture du pore de transition de perméabilité mitochondrial, un événement provoqué par l’augmentation du stress oxydatif et la défaillance de la chaîne respiratoire, aboutissant au relargage du cytochrome c et à l’activation des caspases [32].
Inhibition de la GSK-3β et thérapeutique de la MP
Il existe peu de données sur une éventuelle thérapeutique anti-Parkinson basée sur l’inhibition de la GSK-3β. Néanmoins, un traitement au long cours par le lithium protège les neurones dopaminergiques des souris traitées par le MPTP et préserve le contenu cérébral en dopamine [33]. De plus, tout comme dans la MA, la dérégulation de l’activité de la GSK-3β est un élément central dans la pathogenèse de la MP, puisqu’elle agit sur le dysfonctionnement mitochondrial, la mort neuronale, ainsi que l’agrégation des protéines Tau, α-synucléine et synphiline-1. La GSK-3β apparaît donc comme une cible thérapeutique privilégiée pour prévenir l’initiation et la progression de la maladie.
GSK-3β et autres maladies neurodégénératives
La maladie de Huntington (MH)
La MH est liée à une expansion anormale de polyglutamine dans la protéine huntingtine, causant son agrégation et l’interruption de processus cellulaires nécessaires à la survie neuronale. L’inhibition de la GSK-3β par le lithium ou l’expression d’une forme dominante négative de la protéine protège les neurones contre la mort induite par les polyglutamines grâce à l’élévation du niveau de β-caténine [34]. De plus, le lithium réduirait la quantité des agrégats protéiques intracellulaires. Combiné à la rapamycine, un activateur d’autophagie favorisant la dégradation des agrégats protéiques, le lithium présenterait un fort potentiel thérapeutique dans la MH [35].
La sclérose latérale amyotrophique (SLA)
La SLA est une maladie neurologique qui affecte les neurones moteurs situés dans la corne antérieure de la moelle épinière, du tronc cérébral et du cortex. L’expression de la GSK-3β totale et de sa forme active est augmentée dans les tissus nerveux de patients atteints de SLA. Cette surexpression coïncide avec des dépôts de la protéine Tau hyperphosphorylée [36]. Dans un modèle murin de SLA, l’activité neuronale de la GSK-3β est augmentée, et l’inhibition de son activité empêche la mort cellulaire. De plus, l’inhibition de la GSK-3β par un traitement combiné de valproate et de lithium retarde l’apparition des symptômes, freine la progression de la maladie et prolonge l’espérance de vie de souris transgéniques modèles de la maladie [37].
Encéphalopathies spongiformes subaiguës transmissibles (ESST)
Les ESST ou maladies à prions sont dues à l’accumulation au niveau cérébral d’agrégats essentiellement composés de la protéine prion sous sa forme pathologique. Un peptide constitué de la séquence 106-126aa de la protéine prion est couramment utilisé dans l’étude de la neurodégénérescence liée aux maladies à prions. Dans des cultures primaires de neurones, le peptide 106-126 augmente l’activité de la GSK-3β et induit la mort cellulaire de manière dépendante de l’expression et de l’activité de la kinase [38]. Le lithium a des effets neuroprotecteurs vis-à-vis de la toxicité du peptide et augmente le temps de survie de souris infectées par les prions [39].
Conclusion
Le rôle majeur de la GSK-3β dans les maladies neurodégénératives est irréfutable. Bien que ces pathologies possèdent des origines et des processus moléculaires qui leur sont propres, elles présentent des similitudes dans leur pathogenèse. L’accumulation et l’agrégation de protéines toxiques, le dysfonctionnement du métabolisme cellulaire, l’inflammation et la mort neuronale par apoptose sont des dénominateurs communs de ces pathologies. L’augmentation de l’activité de la GSK-3β par des phosphorylations régulatrices des résidus Ser9 et Tyr216 semble être le point de convergence de nombreuses voies de signalisation pathologiques. D’un point de vue théorique, certaines réserves quant à l’utilisation à long terme d’inhibiteurs de GSK-3β pouvaient initialement être formulées du fait de l’accumulation possible de la b-caténine et des éventuelles conséquences oncogéniques. Néanmoins, de nombreuses expériences ont apporté des conclusions rassurantes à ce sujet. Le lithium, classiquement utilisé dans le traitement des troubles bipolaires et de la dépression, n’augmente pas l’incidence des cas de cancer chez la population traitée, ce qui indique qu’une inhibition modérée au long cours de la GSK-3β n’a pas d’effets secondaires délétères susceptibles de provoquer ce type de pathologie. Les résultats des études cliniques en cours seront déterminants pour savoir si la GSK-3β est une cible thérapeutique prometteuse ou illusoire.
Conflit d’intérêts
L’auteur déclare n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
La β-caténine est un composant de la voie de signalisation Wnt : « en l’absence de Wnt, elle est incluse dans un complexe protéique comprenant l’axine, APC et la kinase GSK3-β (glycogen synthase kinase 3β). Elle est phosphorylée par cette dernière, ce qui entraîne sa dégradation par le protéasome » (tiré de [40]).
En 1912, Fritz H. Lewy relève la présence d’inclusions cellulaires intracytoplasmiques dans le tronc cérébral des patients atteints de la MP. « Ces agrégats - appelés aussi corps de Lewy - exprimant la synucléine et l’ubiquitine dans le corps cellulaire ou les neurites de certains neurones dopaminergiques constituent les stigmates neuropathologiques caractéristiques du processus neurodégénératif de la MP » (tiré de [44]).
« L’α-synucléine est une protéine neuronale de 140 acides aminés, extrêmement conservée chez les vertébrés. Elle est enrichie dans de nombreuses régions cérébrales (néocortex, hippocampe, gyrus denté, bulbe olfactif, striatum, thalamus, cervelet) dans lesquelles elle se concentre au niveau des terminaisons synaptiques. […] Les fibrilles insolubles d’α-synucléine sont un composant majeur des corps de Lewy » (tiré de [42]).
La synphiline-1 est un partenaire de l’α-synucléine et substrat de caspases.
Références
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 2001; 65 : 391–426. [Google Scholar]
- Leroy K, Yilmaz Z, Brion JP. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol 2007; 33 : 43–55. [Google Scholar]
- Brownlees J, Irving NG, Brion JP, et al. Tau phosphorylation in transgenic mice expressing glycogen synthase kinase-3beta transgenes. Neuroreport 1997; 8 : 3251–5. [Google Scholar]
- Engel T, Hernandez F, Avila J, et al. Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci 2006; 26 : 5083–90. [Google Scholar]
- Aplin AE, Gibb GM, Jacobsen JS, et al. In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3beta. J Neurochem 1996; 67 : 699–707. [Google Scholar]
- Qing H, He G, Ly PT, et al. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J Exp Med 2008; 205 : 2781–9. [Google Scholar]
- Su Y, Ryder J, Li B, et al. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry 2004; 43 : 6899–908. [Google Scholar]
- Terwel D, Muyllaert D, Dewachter I, et al. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol 2008; 172 : 786–98. [Google Scholar]
- Checler F, Alves da Costa C, Dumanchin-Njock C, et al. Métabolisme du précurseur du peptide amyloïde et présénilines. Med Sci (Paris) 2002; 18 : 717–24. [Google Scholar]
- Takashima A, Murayama M, Murayama O, et al. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc Natl Acad Sci USA 1998; 95 : 9637–41. [Google Scholar]
- Murayama M, Tanaka S, Palacino J, et al. Direct association of presenilin-1 with beta-catenin. FEBS Lett 1998; 433 : 73–7. [Google Scholar]
- Rockenstein E, Torrance M, Adame A, et al. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci 2007; 27 : 1981–91. [Google Scholar]
- Nakashima H, Ishihara T, Suguimoto P, et al. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol 2005; 110 : 547–56. [Google Scholar]
- Engel T, Goni-Oliver P, Lucas JJ, et al. Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J Neurochem 2006; 99 : 1445–55. [Google Scholar]
- Caccamo A, Oddo S, Tran LX, et al. Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am J Pathol 2007; 170 : 1669–75. [Google Scholar]
- Hu S, Begum AN, Jones MR, et al. GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis 2009; 33 : 193–206. [Google Scholar]
- Jope RS, Bijur GN. Mood stabilizers, glycogen synthase kinase-3beta and cell survival. Mol Psychiatry 2002; 7 (suppl 1) : S35–45. [Google Scholar]
- Gomez-Sintes R, Hernandez F, Bortolozzi A, et al. Neuronal apoptosis and reversible motor deficit in dominant-negative GSK-3 conditional transgenic mice. EMBO J 2007; 26 : 2743–54. [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 2007; 53 : 703–17. [Google Scholar]
- Bell EC, Willson MC, Wilman AH, et al. Differential effects of chronic lithium and valproate on brain activation in healthy volunteers. Hum Psychopharmacol 2005; 20 : 415–24. [Google Scholar]
- Martinez A, Perez DI. GSK-3 inhibitors: a ray of hope for the treatment of Alzheimer’s disease ? J Alzheimers Dis 2008; 15 : 181–91. [Google Scholar]
- Dagda RK, Zhu J, Chu CT. Mitochondrial kinases in Parkinson’s disease: Converging insights from neurotoxin and genetic models. Mitochondrion 2009; 9 : 289–98. [Google Scholar]
- Nagao M, Hayashi H. Glycogen synthase kinase-3beta is associated with Parkinson’s disease. Neurosci Lett 2009; 449 : 103–7. [Google Scholar]
- Wang W, Yang Y, Ying C, et al. Inhibition of glycogen synthase kinase-3beta protects dopaminergic neurons from MPTP toxicity. Neuropharmacology 2007; 52 : 1678–84. [Google Scholar]
- Petit-Paitel A, Brau F, Cazareth J, et al. Involvment of cytosolic and mitochondrial GSK-3beta in mitochondrial dysfunction and neuronal cell death of MPTP/MPP-treated neurons. PLoS One 2009; 4 : e5491. [Google Scholar]
- Arima K, Hirai S, Sunohara N, et al. Cellular co-localization of phosphorylated tau- and NACP/alpha-synuclein-epitopes in lewy bodies in sporadic Parkinson’s disease and in dementia with Lewy bodies. Brain Res 1999; 843 : 53–61. [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, et al. Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol 2002 : 104 : 7–11. [Google Scholar]
- Duka T, Duka V, Joyce J N, et al. Alpha-synuclein contributes to GSK-3(beta)-catalyzed Tau phosphorylation in Parkinson’s disease models. Faseb J 2009; 23 : 2820–30. [Google Scholar]
- Ribeiro CS, Carneiro K, Ross CA, et al. Synphilin-1 is developmentally localized to synaptic terminals, and its association with synaptic vesicles is modulated by alpha-synuclein. J Biol Chem 2002; 277 : 23927–33. [Google Scholar]
- Coux O, Piechaczyk M. Le système ubiquitine/protéasome : un ensemble (de) complexe(s) pour dégrader les protéines. Med Sci (Paris) 2000; 16 : 623–9. [Google Scholar]
- Avraham E, Szargel R, Eyal A, et al. Glycogen synthase kinase 3beta modulates synphilin-1 ubiquitylation and cellular inclusion formation by SIAH: implications for proteasomal function and Lewy body formation. J Biol Chem 2005 : 280 : 42877–86. [Google Scholar]
- Park SS, Zhao H, Mueller RA, et al. Bradykinin prevents reperfusion injury by targeting mitochondrial permeability transition pore through glycogen synthase kinase 3beta. J Mol Cell Cardiol 2006; 40 : 708–16. [Google Scholar]
- Youdim MB, Arraf Z. Prevention of MPTP (N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) dopaminergic neurotoxicity in mice by chronic lithium: involvements of Bcl-2 and Bax. Neuropharmacology 2004; 46 : 1130–40. [Google Scholar]
- Carmichael J, Sugars KL, Bao YP, et al. Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J Biol Chem 2002; 277 : 33791–8. [Google Scholar]
- Sarkar S, Ravikumar B, Floto RA, et al. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 2009; 16 : 46–56. [Google Scholar]
- Yang W, Leystra-Lantz C, Strong MJ. Upregulation of GSK3beta expression in frontal and temporal cortex in ALS with cognitive impairment (ALSci). Brain Res 2008; 1196 : 131–9. [Google Scholar]
- Feng HL, Leng Y, Ma CH, et al. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 2008; 155 : 567–72. [Google Scholar]
- Perez M, Rojo AI, Wandosell F, et al. Prion peptide induces neuronal cell death through a pathway involving glycogen synthase kinase 3. Biochem J 2003; 372 : 129–36. [Google Scholar]
- Heiseke A, Aguib Y, Riemer C, et al. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem 2009; 109 : 25–34. [Google Scholar]
- Andreu P, Perret C, Romagnolo B. Wnt et cellules souches intestinales : des liaisons dangereuse Med Sci (Paris) 2006; 22 : 693–5. [Google Scholar]
- Clavaguera F, Goedert M, Tolnay M. Induction et propagation de la pathologie par la protéine tau chez un modèle murin de la maladie d’Alzheimer. Med Sci (Paris) 2010; 26 : 123–4. [Google Scholar]
- Corti O, Brice A. La maladie de Parkinson: que nous apprennent les gènes responsables des formes familiales ? Med Sci (Paris) 2003; 19 : 613–9. [Google Scholar]
- Langui D, Lachapelle F, Duyckaerts C. Modèles animaux des maladies neuro-dégénératives. Med Sci (Paris) 2007; 23 : 180–6. [Google Scholar]
- Lelan F, Damier P. Les neurones dopaminergiques greffés dans la maladie de Parkinson sont-il à leur tour atteints par le processus dégénératif ? Med Sci (Paris) 2009; 25 : 15–6. [Google Scholar]
Liste des figures
|
Figure 1. Représentation schématique du rôle central de la GSK-3β dans les voies neurodégénératives aboutissant à la mort neuronale. Diverses neurotoxines, dont le peptide amyloïde Aβ impliqué dans la maladie d’Alzheimer, le MPTP et la 6-hydroxydopamine (6-OHDA) dont les effets miment la maladie de Parkinson, activent, directement ou indirectement, l’activité de la GSK-3β en augmentant la phosphorylation du résidu tyrosine 216 (pTyr216) et en inhibant la phosphorylation du résidu sérine 9 (pSer9). L’activation de la GSK-3 β par le peptide amyloïde a pour conséquence l’hyperphosphorylation de la protéine Tau à l’origine des dégénérescences neurofibrillaires dans la maladie d’Alzheimer ainsi que l’augmentation de la production du peptide amyloïde lui-même, ce qui aboutit à la formation de plaques séniles. Les deux lésions histopathologiques caractéristiques de la maladie d’Alzheimer sont les agrégats de protéine Tau hyperphosphorylée et les plaques séniles. L’activation de la GSK-3β dépendante de l’α-synucléine par le MPTP et la 6-OHDA entraîne l’hyperphosphorylation de la protéine Tau et son agrégation (détectée dans le cerveau de nombreux patients parkinsoniens), la phosphorylation et l’agrégation de l’α-synucléine au sein des corps de Lewy et la baisse de l’activité mitochondriale dans les neurones touchés. Tous ces phénomènes aboutissent à la mort neuronale dans des modèles cellulaires et animaux de la maladie de Parkinson. |
| Dans le texte | |
|
Figure 2. Représentation schématique de l’implication de la GSK-3β dans les voies cellulaires mises en jeu dans la maladie d’Alzheimer. Au cours de la maladie d’Alzheimer, la GSK-3β influence le clivage de la protéine précurseur de l’amyloïde (APP) par la γ-sécrétase et favorise ainsi la production du peptide amyloïde Aβ qui s’accumule dans l’espace extracellulaire et s’agrège sous la forme de plaques séniles. En retour, l’Aβ active la GSK-3β, entraînant l’hyperphosphorylation de la protéine Tau qui se dissocie des microtubules et provoque l’apparition de dégénérescences neurofibrillaires. La GSK-3β interagit également avec un membre du complexe γ-sécrétase, la préséniline-1, qui elle-même interagit avec la β-caténine. La β-caténine est phosphorylée par la GSK-3β, ce qui induit sa dégradation par le protéasome. Enfin, la GSK-3β activée favorise la mort neuronale en activant la voie apoptotique impliquant le relargage du cytochrome c, l’activation de la cascade des caspases et la fragmentation de l’ADN. |
| Dans le texte | |
|
Figure 3. Représentation schématique de l’implication de la GSK-3β dans les voies cellulaires impliquées dans la maladie de Parkinson. Dans les modèles de maladie de Parkinson par intoxication à la neurotoxine MPTP, la GSK-3β est activée par l’intermédiaire de l’α-synucléine. En retour, la GSK-3β favorise l’agrégation de l’α-synucléine et de la synphiline-1 au sein des corps de Lewy. La 6-hydroxydopamine (6-OHDA) induit l’activation, directe ou indirecte, de la GSK-3β. La GSK-3β activée est responsable de l’hyperphosphorylation de la protéine Tau. Enfin, sous l’effet des neurotoxines, la GSK-3β activée provoque la chute du potentiel membranaire mitochondrial (Ψm), entraînant l’ouverture du pore de transition de perméabilité mitochondrial, ce qui aboutit au relargage du cytochrome c dans le cytosol, à l’activation de la cascade des caspases, à la fragmentation de l’ADN et à la mort neuronale par apoptose. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.