")
")
| Issue |
Med Sci (Paris)
Volume 24, Number 11, Novembre 2008
|
|
|---|---|---|
| Page(s) | 912 - 915 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20082411912 | |
| Published online | 15 November 2008 | |
Lost after translation
Lost after translation
Professeur émérite à la Faculté de Médecine Paris Descartes, Université Paris Descartes, Institut Cochin, 24, rue du Faubourg Saint-Jacques, 75014 Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Un allèle faux-sens se comportant comme un allèle nul
Le numéro de mars 2008 du journal Human Molecular Genetics contient deux articles [1, 2] portant sur la pathogénicité de la mutation fauxsens R77C dans le gène de l’alphasarcoglycane (SGCA). La pathologie de ce gène entraîne chez l’homme une dystrophie musculaire progressive de sévérité variable [5–8]. Rappelons que les 4 sarcoglycanes (α, β, γ et δ−SG) sont des protéines monomériques transmembranaires à un seul passage et O-glycosylées sur leur versant extracellulaire. Elles s’associent pour former le complexe des sarcoglycanes (SG) faisant lui-même partie du complexe DGC (Dystrophin glycoprotein complex) (Figure 1). L’ensemble assure l’amarrage du cytosquelette de la cellule musculaire aux protéines de la matrice extracellulaire, via l’axe dystrophine-dystroglycanes [9,10]. Le mode d’assemblage de cet édifice sarcolemmique a été éclairci [11, 12]. Les mutations touchant n’importe lequel des 4 gènes du complexe SG provoquent un démantèlement de tout l’édifice, et il s’ensuit une dystrophie musculaire des ceintures de transmission autosomique récessive, désignée LGMD2D, E, C, F selon le gène en cause [8, 13]. On ne dispose encore d’aucun traitement pour ces myopathies. C’est la pathologie du gène SGCA (LGMD2D, OMIM 608099) qui est la plus fréquente [14]. Chez la souris, le knock-out du gène Sgca a déjà été effectué (souris SgcaNull/Null), de même que celui des 3 autres gènes de sarcoglycane, réalisant dans tous les cas un phénotype de dystrophie musculaire sévère [15, 16]. Or, en pathologie humaine, la majorité des mutations du gène SGCA sont des faux-sens affectant le domaine extracellulaire [17]. Parmi celles-ci, la mutation c.229C>T au niveau d’un CpG de l’exon 3, qui induit le remplacement en position 77 d’une arginine par une cystéine (R77C), est remarquablement récurrente puisqu’on la trouve sous nos latitudes chez plus d’un tiers des malades [6, 17]. Chez ceux-ci, le transcrit muté est en quantité normale, mais au niveau du sarcolemme la protéine est absente ou très diminuée, ce qui suggère une instabilité du produit fini ou une anomalie de maturation. Pour préciser la pathogénie et disposer d’un modèle thérapeutique il était donc logique de créer par knock-in une souris portant la mutation faux-sens prédominante dans les α-sarcoglycanopathies humaines.
 |
Figure 1. Schéma simplifié de la dystrophin connection. Seules sont représentés les partenaires les plus directs. Les arborescences symbolisent les restes O-glycosylés. La mutation R77C porte sur le gène de l’α-sarcoglycane (en rouge) (modifié d’après [36]). |
Les souris SgcH77C/H77C sont asymptomatiques
L’alpha-sarcoglycane est très conservée au cours de l’évolution, mais chez la souris l’acide aminé en position 77 est une histidine au lieu d’une arginine, ce qui n’affecte pas la charge positive du résidu. Les deux équipes mentionnées ci-dessus [1, 2] ont effectué avec succès le remplacement du résidu His par le résidu Cys dans le gène murin. À leur grande surprise les souris obtenues (SgcaH77C/H77C) ne manifestent aucun symptôme clinique ou histologique de dystrophie musculaire, et la protéine mutée est normalement exprimée et positionnée au niveau du sarcolemme. Cela contraste avec la pathologie généralement sévère observée chez les malades homozygotes pour la mutation R77C [6, 7].
La protéine α-SG humaine R77C est bel et bien fabriquée mais elle est perdue en cours de route
L’équipe d’Isabelle Richard [2] en apporte la preuve expérimentale grâce à des expériences de complémentation hétérospécifique effectuées ex vivo. Des rétinoblastes embryonnaires humains, normalement dépourvus de sarcoglycanes (lignée HER911), sont d’abord cotransfectés avec des plasmides contenant l’ADNc des β-, γ- et δ-SG humaines, puis complémentés avec un plasmide contenant l’ADNc de l’α-SG humaine, dans sa version normale, ou mutée R77C. La version non mutée de l’ADNc de l’α-SG permet d’obtenir une reconstitution du complexe des quatre SG au niveau de la membrane plasmique. Cette restauration n’est pas obtenue avec la version R77C. Mais, si les cellules sont perméabilisées pour permettre l’accès des anticorps aux antigènes présents à l’intérieur de la cellule, on retrouve la protéine R77C dans l’appareil sécrétoire intracellulaire où elle s’est accumulée. Ce résultat suggère une physio-pathologie par anomalie de maturation empêchant la protéine d’arriver à sa destination finale. En fait, la preuve d’une rétention de la protéine R77C immature dans le compartiment du réticulum endoplasmique (ER) avait déjà été bien documentée par Draviam et al. [18], grâce au marquage moléculaire par la GFP de la protéine normale et mutée, dans un autre système de cellules humaines (HK293). Ce travail avait en outre permis de reconstituer le parcours effectué par l’allèle normal depuis le ribosome jusqu’au sarcolemme via le système ER, puis le Golgi et le système de transport microtubulaire [18]. Fait intéressant, la protéine α-SG n’a pas besoin de ses partenaires β, γ et δ-SG pour migrer à la membrane, mais elle en a besoin pour s’y maintenir de façon stable, ce qui confirme que la mutation R77C n’affecte pas les interactions avec les autres membres du complexe [18].
Sauvetage de l’allèle R77C par relaxation du contrôle de qualité ERAD
Rappelons que la maturation posttraductionnelle des glycoprotéines exportées a lieu dans le réticulum endoplasmique, et permet aux chaînes polypeptidiques naissantes d’acquérir une configuration finale correcte, seule forme exportable vers sa destination subcellulaire finale (ici le sarcolemme). Le processus comporte un contrôle de qualité au cours duquel les protéines n’ayant pas acquis une configuration normale sont détectées, reconduites à la frontière ER/cytosol, et expulsées par rétrotranslocation vers le cytosol où elles sont détruites dans le protéasome 26S [34, 35]. Ce système, appelé ERAD pour endoplasmic reticulum associated degradation est d’une grande complexité, et met en jeu au moins 15 protéines différentes. Il a donné lieu à une abondante littérature à partir des années 1980 (pour revues, voir [19, 20]). La Figure 2 et l’Encadré en schématisent les grands traits. Ce contrôle de qualité est très tâtillon, car moins de la moitié des protéines normales parviennent à sortir par la bonne porte [34, 35]. On sait depuis longtemps manipuler spécifiquement ces systèmes par des molécules inhibant les enzymes intervenant dans les remaniements de la chaîne oligosaccharidique [21, 22]. Bartoli et al. [2] ont essayé ces agents et ont démontré de manière convaincante que la kifunensine, un inhibiteur spécifique de la mannosidase I [21] (Figure 2) permet de faire réapparaître l’ α-SG R77C à la membrane. Ce résultat spectaculaire a été obtenu non seulement ex vivo dans le système cellulaire reconstitué décrit ci-dessus, mais aussi et surtout in vivo dans un système hétérologue où des souris Sgca−/− reçoivent des injections intra-musculaires d’un transgène α-SG humain vectorisé dans l’AAV (adenoassociated virus). La version non mutée de l’ADNc permet d’obtenir une restauration du complexe SG sarcolemmique. En revanche la version R77C ne permet pas cette complémentation, sauf si après l’injection du vecteur AAV-R77C les souris reçoivent de la kifunensine administrée par voie intra-musculaire. L’effet thérapeutique local est spectaculaire, car l’administration du produit entraîne une reconstitution des complexes protéiques sarcolemmiques, avec un bénéfice cellulaire attesté par le retour de la membrane musculaire à un état d’imperméabilité au bleu Evans. Ces résultats comportent plusieurs preuves de principe très importantes : (1) l’allèle R77C est bel et bien traduit, mais, au cours de sa maturation, la protéine est retenue et détruite par le système ERAD ; (2) la simple inhibition de la mannosidase I suffit à lui laisser un répit nécessaire pour acquérir une configuration acceptable et quitter le compartiment ER par la bonne porte ; (3) la protéine ainsi soustraite à l’ERAD est pleinement fonctionnelle malgré l’anomalie R77C, ce qui est en accord avec le phénotype normal des souris SgcaH77C/H77C . Bien sûr, il reste à comprendre la tolérance du modèle murin vis-à-vis de la mutation 77C sur sa propre α-SG. Mais le problème principal est d’exploiter les potentialités thérapeutiques de cette découverte.
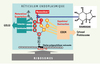 |
Figure 2. La minuterie du système ERAD par la copule N- oligosaccharidique fixée sur la protéine. Les carrés A et B : résidus N-acétylglucosamines. Les cercles C à K : restes mannosyl. Les triangles L à N : restes glucosyl. Les résidus figurés en vert interagissent avec les protéines chaperons d’aide à la configuration (calnexine et calréticuline). Les résidus figurés en jaune interagissent avec les protéines chaperons du complexe EDM reconnaissant et expulsant les chaînes malformées, seulement si l’ablation du résidu mannosyl (cercle rouge I) a eu lieu. Les enzymes ne sont pas indiquées à l’exception de la mannosidase I, spécifiquement inhibée par la kifunensine. |
La pharmacologie à la rescousse des protéines en perdition
La pathogénicité de l’allèle R77C fait entrer la myopathie induite dans la catégorie des « maladies conformationnelles » [23]. On a déjà dénombré plus de 60 maladies dues à une mutation affectant le trafic post-traductionnel [19, 23, 24]. Parmi elles figure notamment la mutation deltaF508 dans le gène CFTR qui affecte 2/3 des malades atteints de mucoviscidose [25]. Il est très probable que les nombreuses autres sarcoglycanopathies par mutations faux-sens [17] ressortissent du même mécanisme. Plus généralement on doit y penser chaque fois qu’une mutation faux-sens avec des transcrits en quantité normale se comporte comme un allèle nul ou très diminué, ce qui est la règle plutôt que l’exception [23]. Il faut savoir que l’accumulation d’une protéine malformée dans l’ER est à l’origine d’un stress aux multiples conséquences sur les voies de transduction du signal, pouvant culminer dans un syndrome dit d’UPR (unfolded protein response) entraînant la mort cellulaire par apoptose ou autophagie [26, 33, 37] (→). De ce point de vue, les allèles porteurs de mutations faux-sens seraient pathogènes à la fois par leur absence au bon endroit (LOF), et par la toxicité induite par l’engorgement du compartiment ER (GOF).
(→) Voir l’article de Éric Chevet, page 899 de ce numéro
Concrètement, ces résultats étant acquis, il va falloir résoudre rapidement le problème de la toxicité de la kifunensine et des autres molécules agissant sur la même cible. En effet à notre connaissance, cet alcaloïde découvert en 1991 dans l’actinomycète Kitasatosporia kifunense, [21] n’a jusqu’ici été utilisé qu’in vitro ou ex vivo [22]. Il est encourageant de noter que dans les expériences rapportées plus haut [2], l’administration par voie intramusculaire n’a pas entraîné chez les souris d’effet néfaste local ou général. Mais si cette stratégie thérapeutique devenait envisageable en clinique, le modèle animal, indispensable pour les analyses précliniques, fait pour le moment défaut. De plus, en provoquant un relâchement de la vigilance du système ERAD on court le risque théorique de laisser échapper non seulement l’allèle désiré mais d’autres espèces malformées dont l’effet pourrait être néfaste. Dans cet ordre d’idée on cherche activement des molécules diffusibles agissant uniquement sur l’allèle pathologique, d’où l’appellation de « chaperon pharmacologique » [24, 27]. Cette spécificité a été obtenue, paradoxalement, avec des inhibiteurs compétitifs qui, à faibles doses, se lient à la cible par une liaison de type enzyme-substrat et l’aident à atteindre la configuration normale [28]. Cette approche s’est avérée efficace, notamment dans les maladies lysosomiales dues à des mutations faux-sens, comme la maladie de Fabry (déficit en α−galactosidase) [29]. La recherche pharmacologique dans ce sens va bon train, notamment grâce à l’élaboration de modèles cellulaires se prêtant à un criblage à haut débit [30]. C’est ainsi qu’on vient de découvrir l’effet chaperon de la pyriméthamine – un antipaludéen déjà sur le marché – qui secourt efficacement des allèles anormaux de la β-hexosaminidase A en perdition post-traductionnelle. D’où l’espoir de pouvoir traiter les formes tardives de maladies de Tay-Sachs ou de Sandhoff [31]. Ainsi, après le concept thérapeutique quelque peu simpliste de thérapie génique par un « ADN-médicament », on arrive à la notion de thérapie génétique fondée sur la connaissance des gènes (gene-based therapy), de leurs mutations et des différents niveaux de dysfonctions qu’elles engendrent. La pharmacopée des protéines mutées a un bel avenir [32].

Références
- Kobuke K, Piccolo F, Garringer KW, et al. A common disease-associated missense mutation in alphasarcoglycan fails to cause muscular dystrophy in mice. Hum Mol Genet 2008; 17 : 1201–13. [Google Scholar]
- Bartoli M, Gicquel E, Barrault L, et al. Mannosidase I inhibition rescues the human [alpha]-sarcoglycan R77C recurrent mutation. Hum Mol Genet 2008; 17 : 1214–21. [Google Scholar]
- Roberds SL, Anderson RD, Ibraghimov-Beskrovnaya O, Campbell KP. Primary structure and muscle-specific expression of the 50-kDa dystrophin-associated glycoprotein (adhalin). J Biol Chem 1993; 268 : 23739–42. [Google Scholar]
- Roberds SL, Leturcq F, Allamand V, et al. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell 1994; 78 : 625–33. [Google Scholar]
- Piccolo F, Roberds SL, Jeanpierre M, et al. Primary adhalinopathy : a common cause of autosomal recessive muscular dystrophy of variable severity. Nat Genet 1995; 10 : 243–5. [Google Scholar]
- Carrié A, Piccolo F, Leturcq F, et al. Mutational diversity and hot spots in the alpha-sarcoglycan gene in autosomal recessive muscular dystrophy (LGMD2D). J Med Genet 1997; 34 : 470–5. [Google Scholar]
- Eymard B, Romero NB, Leturcq F, et al. Primary adhalinopathy (alpha-sarcoglycanopathy) : clinical, pathological and genetic correlation in twenty patients with autosomal recessive muscular dystrophy. Neurology 1997; 4 : 1227–34. [Google Scholar]
- Kaplan JC, Beckmann JS, Fardeau M. Limb girdle muscular dystrophies. In : Karpati G, Hilton-Jones D, Griggs R, eds. Disorders of voluntary muscle, chapter 20, 7e ed. Cambridge : Cambridge University Press, 2001 : 433–63. [Google Scholar]
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991; 66 : 1121–31. [Google Scholar]
- Ozawa E, Mizuno Y, Hagiwara Y, et al . Molecular and cell biology of the sarcoglycan complex. Muscle Nerve 2005; 32 : 563–76. [Google Scholar]
- Holt K, Campbell K. Assembly of the sarcoglycan complex. Insights for muscular dystrophy. J Biol Chem 1998; 23 : 34667–70. [Google Scholar]
- Allikian MJ, McNally EM. Processing and assembly of the dystrophin glycoprotein complex. Traffic 2007; 8 : 177–83. [Google Scholar]
- Lim L, Campbell K. The sarcoglycan complex in limbgirdle muscular dystrophy. Curr Opin Neurol 1998; 11 : 443–52. [Google Scholar]
- Trabelsi M, Kavian N, Daoud F, et al. Revised spectrum of mutations in sarcoglycanopathies. Eur J Hum Genet 2008; 16 : 793–803. [Google Scholar]
- Duclos F, Straub V, Moore SA, et al. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J Cell Biol 1998; 142 : 1461–71. [Google Scholar]
- Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex : an overview of current mouse models. Curr Opin Genet Dev 2002; 12 : 349–61. [Google Scholar]
- LOVD. Leiden Muscular Dystrophy pages. http://www. dmd.nl/nmdb/home.php [Google Scholar]
- Draviam RA, Wang B, Shand SH, et al. Alphasarcoglycan is recycled from the plasma membrane in the absence of sarcoglycan complex assembly. Traffic 2006; 7 : 793–810. [Google Scholar]
- Hebert DN , Molinari M. In and out of the ER : protein folding, quality control, degradation, and related human diseases. Physiol Rev 2007; 87 : 1377–408. [Google Scholar]
- Roth J, Yam GH, Fan J, et al. Protein quality control : the who’s who, the where’s and therapeutic escapes. Histochem Cell Biol 2008; 129 : 163–77. [Google Scholar]
- Elbein AD, Kerbacher JK, Schwartz CJ, Sprague EA. Kifunensine inhibits glycoprotein processing and the function of the modified LDL receptor in endothelial cells. Arch Biochem Biophys 1991; 288 : 177–84. [Google Scholar]
- Tokunaga F, Brostrom C, Koide T, Arvan P. Endoplasmic reticulum (ER)-associated degradation of misfolded N-linked glycoproteins is suppressed upon inhibition of ER mannosidase I. J Biol Chem 2000; 275 : 40757–64. [Google Scholar]
- Gregersen N, Bross P, Vang S, Christensen JH. Protein misfolding and human disease. Annu Rev Genomics Hum Genet 2006; 7 : 103–24. [Google Scholar]
- Aridor M. Visiting the ER : the endoplasmic reticulum as a target for therapeutics in traffic related diseases. Adv Drug Deliv Rev 2007; 59 : 759–81. [Google Scholar]
- French PJ, van Doorninck JH, Peters RH, et al. A delta F508 mutation in mouse cystic fibrosis transmembrane conductance regulator results in a temperature-sensitive processing defect in vivo. J Clin Invest 1996; 98 : 1304–12. [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8 : 519–29. [Google Scholar]
- Bernier V, Lagacé M, Bichet DG, Bouvier M. Pharmacological chaperones : potential treatment for conformational diseases. Trends Endocrinol Metab 2004; 15 : 222–8. [Google Scholar]
- Fan JQ. A counterintuitive approach to treat enzyme deficiencies : use of enzyme inhibitors for restoring mutant enzyme activity. Biol Chem 2008; 389 : 1–11. [Google Scholar]
- Hamanaka R, Shinohara T, Yano S, et al. Rescue of mutant alpha-galactosidase A in the endoplasmic reticulum by 1-deoxygalactonojirimycin leads to trafficking to lysosomes. Biochim Biophys Acta 2008; 1782 : 408–13. [Google Scholar]
- Pey AL, Ying M, Cremades N, et al. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J Clin Invest 2008; 118 : 2858–67. [Google Scholar]
- Tropak MB, Mahuran D. Lending a helping hand, screening chemical libraries for compounds that enhance beta-hexosaminidase; A activity in GM2 gangliosidosis cells. FEBS Journal 2007; 274 : 4951–61. [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science 2008; 319 : 916–9. [Google Scholar]
- Foufelle F, Ferré P. La réponse UPR : son rôle physiologique et physiopathologique. Med Sci (Paris) 2007; 23 : 291–6. [Google Scholar]
- Yon-Kahn J. Repliement des protéines : études in vitro. Med Sci (Paris) 2005; 21 : 601–7. [Google Scholar]
- Goldberg ME. Le repliement des protéines : seconde traduction du message génétique. Med Sci (Paris) 2005; 21 : 563–6. [Google Scholar]
- Kaplan JC, Delpech M. Biologie moléculaire et médecine, 3e ed. Collection De la biologie à la clinique. Paris : Flammarion Médecine-Sciences, 2007 : 820 p. [Google Scholar]
- Chevet E. Inflammation intestinale et stress du réticulum endoplasmique : un lien génétique. Med Sci (Paris) 2008; 24 : 899–900. [Google Scholar]
© 2008 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Schéma simplifié de la dystrophin connection. Seules sont représentés les partenaires les plus directs. Les arborescences symbolisent les restes O-glycosylés. La mutation R77C porte sur le gène de l’α-sarcoglycane (en rouge) (modifié d’après [36]). |
| Dans le texte | |
|
Figure 2. La minuterie du système ERAD par la copule N- oligosaccharidique fixée sur la protéine. Les carrés A et B : résidus N-acétylglucosamines. Les cercles C à K : restes mannosyl. Les triangles L à N : restes glucosyl. Les résidus figurés en vert interagissent avec les protéines chaperons d’aide à la configuration (calnexine et calréticuline). Les résidus figurés en jaune interagissent avec les protéines chaperons du complexe EDM reconnaissant et expulsant les chaînes malformées, seulement si l’ablation du résidu mannosyl (cercle rouge I) a eu lieu. Les enzymes ne sont pas indiquées à l’exception de la mannosidase I, spécifiquement inhibée par la kifunensine. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.