")
")
| Issue |
Med Sci (Paris)
Volume 23, Number 6-7, Juin-Juillet 2007
|
|
|---|---|---|
| Page(s) | 575 - 578 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20072367575 | |
| Published online | 15 juin 2007 | |
Neutral lipid storage diseases et déficits en ATGL (adipose triglycéride lipase) et CGI-58/ABHD5 (α-β hydrolase domain-containing 5) : myopathie, ichtyose, mais pas d’obésité
Neutral lipid storage diseases and ATGL (adipose triglyceride lipase) and CGI-58 deficiency: myopathy, ichthyosis, but no obesity
1
CNG, Centre National de Génotypage, 2, rue Gaston-Crémieux CP 5721, 91057 Évry Cedex, France
2
Inserm U466, Université Paul Sabatier, IFR-31, CHU Rangueil, 1, avenue du Pr-Jean-Poulhès TSA 50032, 31059 Toulouse Cedex, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Dans les cellules eucaryotes, les réserves énergétiques sont constituées par des lipides (triglycérides ou triacylglycérols) et des polysaccharides (glycogène chez les animaux et amidon chez les plantes). Chez les animaux, les triglycérides (triesters d’acides gras et de glycérol) constituent la principale réserve énergétique de l’organisme. Ces triglycérides peuvent être stockés sous forme de gouttelettes lipidiques cytoplasmiques (lipid droplets, formées de triglycérides et d’esters de cholestérol) dans les adipocytes, mais aussi dans toutes les cellules de l’organisme.
Les triglycérides cytoplasmiques sont synthétisés par chaque cellule à partir d’acides gras exogènes (d’origine alimentaire, ou hépatique, et transportés par des lipoprotéines qui libèrent les acides gras lors de leur lipolyse), et endogènes (biosynthétisés par les cellules). Cette biosynthèse se fait à partir d’acyl-CoA et de sn-glycérol-3-phosphate qui donnent du 1,2-diacylglycérol-3-phosphate servant à la synthèse de phospholipides et des triacylglycérols. Ces triglycérides des réserves peuvent être dégradés par divers types de lipases qui hydrolysent des liaisons esters et libèrent progressivement les acides gras [1, 2]. Ceux-ci peuvent être utilisés par la cellule elle-même, pour la production énergétique, ou pour la synthèse de lipides complexes membranaires. Ils peuvent en outre être libérés par la cellule (c’est le cas du tissu adipeux) et transportés par le sang (par l’albumine principalement) vers d’autres cellules utilisatrices (Figure 1).
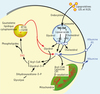 |
Figure 1. Compartiments métaboliques cellulaires des triglycérides (TAG) et des acides gras (AG) [1]. Les lysosomes représentent un premier compartiment : les lipoprotéines (LDL, VLDL) internalisées par la voie des récepteurs apoB/E et l’endocytose clathrine-dépendante sont adressées vers les lysosomes où la lipase acide lysosomale dégrade les triglycérides et les esters de cholestérol, en libérant glycérol, cholestérol et acides gras qui sont ensuite transférés dans le cytoplasme. Les gouttelettes lipidiques cytoplasmiques constituent l’autre compartiment subcellulaire contenant des triglycérides qui sont dégradés par l’ATGL qui est ubiquitaire et activée par CGI-58 [2, 8, 9]. À noter que dans le tissu adipeux, la lipase hormono-sensible joue également un rôle majeur dans la dégradation des triglycérides cytoplasmiques [2]. Les acides gras cytoplasmiques proviennent de plusieurs sources: de la captation des acides gras libres extracellulaires (transportés par l’albumine), et de la lipolyse des lipides cellulaires. Ces acides gras libres cellulaires peuvent être libérés dans le milieu extracellulaire ou bien être convertis en acyl-CoA pour être utilisés pour la production énergétique dans la mitochondrie ou pour synthétiser des lipides cellulaires, lipides complexes membranaires (phospholipides et sphingolipides) et triglycérides stockés dans les gouttelettes lipidiques cytoplasmiques). |
Plusieurs maladies génétiques rares sont associées à des accumulations de lipides neutres cellulaires (sous forme de gouttelettes lipidiques cytoplasmiques ou dans les lysosomes) [1], et sont dues soit à un déficit en lipase, soit au stockage d’acides gras excédentaires (Figure 2).
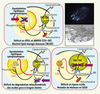 |
Figure 2. Mécanismes physiopathologiques et compartiments subcellulaires d’accumulation des triglycérides (TAG). Dans les NLSD (en haut) et dans des déficits d’utilisation mitochondriale des acides gras (en bas à gauche), les lipides sont localisés dans des gouttelettes lipidiques cytoplasmiques (visibles en microscopie à fluorescence dans des cellules de NLSDM cultivées en présence d’acides gras fluorescents sous forme de compartiments, et en microscopie électronique à transmission) (en haut à droite). Dans les déficits en lipase acide (maladie de Wolman et CESD), les triglycérides et des esters de cholestérol s’accumulent dans les lysosomes. |
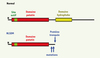 |
Figure 3. Domaines fonctionnels de la lipase PNPLA2/ATGL chez le sujet normal et les patients atteints de NLSD avec myopathie (NLSDM) [5]. |
Le déficit en lipase acide lysosomale (codée par le gène LIPA, localisé en 10q23.2-q23.3) provoque une accumulation de triglycérides et d’esters de cholestérol dans les lysosomes de tissus de patients atteints soit de maladie de Wolman (OMIM 278000), forme grave rapidement mortelle chez l’enfant, soit de CESD (cholesterol ester storage disease) évoluant plus lentement et compatible avec une survie prolongée jusqu’à un âge adulte avancé (la gravité de la maladie est corrélée à la sévérité du déficit fonctionnel de la lipase acide).
Diverses maladies avec déficit mitochondrial du métabolisme des acides gras (déficit systémique en carnitine (gène SLC22A5, localisé en 5q31.1 - OMIM 603377 et 212140), déficit en carnitine musculaire (OMIM 212160), déficit en carnitine-palmitoyl transférase II (CPT2 localisé en 1p32, OMIM 600650), déficit en enzymes de dégradation des acides gras ou de la chaîne respiratoire entre autre…) associent une symptomatologie musculaire, neurologique… souvent grave, et une accumulation de triglycérides cytoplasmiques (mais sans ichtyose).
Enfin des déficits de dégradation des triglycérides cytoplasmiques peuvent aboutir à une accumulation de triglycérides sous forme de gouttelettes lipidiques : ces maladies sont désignées sous le vocable de maladies de stockage de lipides neutres ou neutral lipid storage disease (NSLD).
Les NLSD constituent un groupe de maladies hétérogènes sur le plan clinique et génétique. La symptomatologie clinique comporte une ichtyose (inconstante), un syndrome myopathique, ainsi que d’autres signes inconstants (Tableau I). Elle s’accompagne toujours d’une accumulation multisystémique (dans la plupart des tissus de l’organisme) de triglycérides, en particulier dans les leucocytes qui contiennent des vacuoles lipidiques (anomalie de Jordans) [3]. Les tests biochimiques (après avoir exclu un déficit en carnitine et une pathologie mitochondriale comme le déficit de β-oxydation des acides gras à longue chaîne), montrent un déficit du catabolisme des triglycérides cytoplasmiques qui évoque un déficit du système de la lipase cytoplasmique [1].
Phénotype des patients atteints de NLSD avec myopathie (NLSDM) et de NLSD avec ichtyose (NLSDI ou syndrome de Chanarin-Dorfman, CDS). * Symptômes les plus fréquents ou constants dans la NLSD (donc communs aux deux entités, NLSDM et NLSDI). ** Symptôme (ichtyose) et génétique (anomalies génétiques, mutations dans la séquence) discriminants entre les 2 maladies. Ni : non indiqué. Nombre de cas bien identifiés et publiés : 3 cas de NLSDM ; environ 50 cas de NLSDI/CDS. CGI-58 : comparative gene identification 58 = ABHD5 (α-β hydrolase domain-containing 5). ATGL : adipose triglycéride lipase. 1 Enzymes anormales (muscle [CPK] et foie [SGOT, SGPT, γ-GT, LDH, AP]). 2 Anomalies de l'électroencéphalographie, divers.
Nous avons récemment identifié les gènes [4, 5] impliqués dans deux types de NLSD autosomiques récessives présentant des tableaux cliniques sensiblement différents et nommées NLSD avec ichtyose ou NLSDI) correspondant au syndrome de Chanarin-Dorfman [6, 7] et NLSD avec myopathie (NLSDM [5]. Un premier gène nommé PNPLA2 (localisé en 11p15.5 chez l’homme) code une lipase nommée adipose triglyceride lipase (ATGL) [5, 8], le second, ABHD5 ou CGI-58 (localisé en 3p21) codant une protéine activatrice de l’ATGL [4, 9].
-
NLSD avec ichtyose (NLSDI) et ABHD5 (OMIM 275630 et 604780)
Nous avons proposé de réserver la dénomination NLSD avec ichtyose (NLSDI) ou syndrome de Chanarin-Dorfman (CDS) au sous-groupe de NLSD présentant une ichtyose associée à un cortège de symptômes recensés dans le Tableau I, et un déficit de dégradation des triglycérides cytoplasmiques. La NLSDI est due à des mutations de ABHD5/CGI-58 (sans mutation de PNPLA2/ATGL) [4, 5].
-
NLSD avec myopathie (NLSDM) et PNPLA2/ATGL (OMIM 610717)
Pour un sous-groupe de 3 patients atteints de NLSD avec myopathie modérée (NLSDM), mais sans ichtyose et sans mutation de ABHD5/CGI-58, nous avons recherché des mutations de PNPLA2 qui code l’ATGL, lipase exprimée dans tous les tissus et activée par CGI-58 [2, 8, 9]. Son déficit chez la souris induit une accumulation de triglycérides dans les muscles et la plupart des tissus, et une insuffisance cardiaque sévère [10]. L’analyse des mutations observées chez ces 3 patients par séquençage de la région codante et des jonctions exon-introns a révélé quatre mutations différentes [5]. Trois d’entre elles aboutissent à des protéines tronquées, avec perte du domaine hydrophobe de liaison aux lipides, mais avec conservation du domaine « patatin » qui contient le site actif de la lipase. Ces anomalies structurales prédites sont cohérentes avec les propriétés de la lipase mutée qui conserve une activité enzymatique, mais est incapable de se lier aux gouttelettes de triglycérides. La conséquence est un déficit de dégradation des triglycérides cytoplasmiques, qui est mimé par des siARN dirigés contre l’ATGL [5].
Enfin, il est intéressant de constater qu’aucun des 3 cas de déficit en ATGL ne présente d’obésité. Ces données montrent que, chez l’homme, l’ATGL n’est pas indispensable pour la dégradation des triglycérides dans le tissu adipeux et qu’un déficit d’ATGL n’est probablement pas une cause d’obésité.
Références
- Salvayre R, Negre A, Radom J, Douste-Blazy L. Independence of triacylglycerol-containing compartments in cultured fibroblasts from Wolman disease and multisystemic lipid storage myopathy. FEBS Lett 1989; 250 : 35–9. [Google Scholar]
- Zechner R, Strauss JG, Haemmerle G, et al. Lipolysis: pathway under construction. Curr Opin Lipidol 2005; 16 : 333–40. [Google Scholar]
- Jordans GH. The familial occurrence of fat containing vacuoles in the leukocytes diagnosed in two brothers suffering from dystrophia musculorum progressiva (ERB.). Acta Med Scand 1953; 145 : 419–23. [Google Scholar]
- Lefèvre C, Jobard F, Caux F, et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet 2001; 69 : 1002–12. [Google Scholar]
- Fischer J, Lefevre C, Morava E, et al. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet 2007; 39 : 28–30. [Google Scholar]
- Dorfman ML, Hersko C, Eisenberg S, Sagher F. Ichthyosiform dermatosis with systemic lipidosis. Arch Dermatol 1974; 110 : 261–6. [Google Scholar]
- Chanarin I. Patel A, Slavin G, et al. Neutral-lipid storage disease: a new disorder of lipid metabolism. Br Med J 1975; 1 : 553–5. [Google Scholar]
- Zimmermann R, Strauss JG, Haemmerle G, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004; 306 : 1383–6. [Google Scholar]
- Lass A, Zimmermann R, Haemmerle G, et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab 2006; 3 : 309–19. [Google Scholar]
- Haemmerle G Lass A, Zimmermann R, et al.Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006; 312 : 734–7. [Google Scholar]
© 2007 médecine/sciences - Inserm / SRMS
Liste des tableaux
Phénotype des patients atteints de NLSD avec myopathie (NLSDM) et de NLSD avec ichtyose (NLSDI ou syndrome de Chanarin-Dorfman, CDS). * Symptômes les plus fréquents ou constants dans la NLSD (donc communs aux deux entités, NLSDM et NLSDI). ** Symptôme (ichtyose) et génétique (anomalies génétiques, mutations dans la séquence) discriminants entre les 2 maladies. Ni : non indiqué. Nombre de cas bien identifiés et publiés : 3 cas de NLSDM ; environ 50 cas de NLSDI/CDS. CGI-58 : comparative gene identification 58 = ABHD5 (α-β hydrolase domain-containing 5). ATGL : adipose triglycéride lipase. 1 Enzymes anormales (muscle [CPK] et foie [SGOT, SGPT, γ-GT, LDH, AP]). 2 Anomalies de l'électroencéphalographie, divers.
Liste des figures
|
Figure 1. Compartiments métaboliques cellulaires des triglycérides (TAG) et des acides gras (AG) [1]. Les lysosomes représentent un premier compartiment : les lipoprotéines (LDL, VLDL) internalisées par la voie des récepteurs apoB/E et l’endocytose clathrine-dépendante sont adressées vers les lysosomes où la lipase acide lysosomale dégrade les triglycérides et les esters de cholestérol, en libérant glycérol, cholestérol et acides gras qui sont ensuite transférés dans le cytoplasme. Les gouttelettes lipidiques cytoplasmiques constituent l’autre compartiment subcellulaire contenant des triglycérides qui sont dégradés par l’ATGL qui est ubiquitaire et activée par CGI-58 [2, 8, 9]. À noter que dans le tissu adipeux, la lipase hormono-sensible joue également un rôle majeur dans la dégradation des triglycérides cytoplasmiques [2]. Les acides gras cytoplasmiques proviennent de plusieurs sources: de la captation des acides gras libres extracellulaires (transportés par l’albumine), et de la lipolyse des lipides cellulaires. Ces acides gras libres cellulaires peuvent être libérés dans le milieu extracellulaire ou bien être convertis en acyl-CoA pour être utilisés pour la production énergétique dans la mitochondrie ou pour synthétiser des lipides cellulaires, lipides complexes membranaires (phospholipides et sphingolipides) et triglycérides stockés dans les gouttelettes lipidiques cytoplasmiques). |
| Dans le texte | |
|
Figure 2. Mécanismes physiopathologiques et compartiments subcellulaires d’accumulation des triglycérides (TAG). Dans les NLSD (en haut) et dans des déficits d’utilisation mitochondriale des acides gras (en bas à gauche), les lipides sont localisés dans des gouttelettes lipidiques cytoplasmiques (visibles en microscopie à fluorescence dans des cellules de NLSDM cultivées en présence d’acides gras fluorescents sous forme de compartiments, et en microscopie électronique à transmission) (en haut à droite). Dans les déficits en lipase acide (maladie de Wolman et CESD), les triglycérides et des esters de cholestérol s’accumulent dans les lysosomes. |
| Dans le texte | |
|
Figure 3. Domaines fonctionnels de la lipase PNPLA2/ATGL chez le sujet normal et les patients atteints de NLSD avec myopathie (NLSDM) [5]. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.