")
")
| Issue |
Med Sci (Paris)
Volume 21, Décembre 2005
Métabolisme glucidolipidique et risque cardiovasculaire: Nouvelle approches
|
|
|---|---|---|
| Page(s) | 53 - 54 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20052111s53 | |
| Published online | 15 novembre 2005 | |
Intérêt et limites de la détermination de l’activité enzymatique de l’α-galactosidase A dans les populations à risque pour la maladie de Fabry
Interest and limits of determination of α-galactosidase A enzymatic activity in population at risk for Fabry disease
1
Groupe de Chimie Analytique de Paris Sud, EA 3343, Laboratoire de Chimie Analytique, Faculté de Pharmacie, 5, rue Jean Baptiste Clément, 92296 Châtenay-Malabry, France
2
Unité Fonctionnelle de Génétique Clinique, Hôpital Européen Georges Pompidou, 20, rue Leblanc, 75015 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La prévalence de la maladie de Fabry est très faible dans la population générale. Toutefois, la détermination de l’activité de l’α-galactosidase A présente un intérêt dans les populations à risqué.
Abstract
The prevalence of Fabry disease is low in the regular population. However the determination of the α-galactosidase A activity is of interest in the populations at risk.
© 2005 médecine/sciences - Inserm / SRMS
La maladie de Fabry est une maladie génétique liée au chromosome X qui se caractérise par l’accumulation de globotriaosylcéramide (Gb3) et galabiosylcéramide (Ga2) à la suite d’une mutation du gène GLA codant pour l’α-galactosidase A. Le dosage de l’activité enzymatique de l’enzyme déficiente permet le diagnostic de la maladie chez les hommes hémizygotes (activité résiduelle toujours inférieure à 20 % de la normale). En revanche, le diagnostic chez les femmes hétérozygotes est moléculaire, par l’identification de la mutation du gène GLA. En effet, la détermination de l’activité enzymatique chez les hétérozygotes peut conduire à des résultats très variables, allant théoriquement d’une activité nulle à normale, en fonction de l’inactivation fonctionnelle aléatoire, dans chaque cellule d’un organisme féminin, de l’un des deux chromosomes X [1].
Chez les hétérozygotes dont les signes cliniques sont en général plus tardifs et moins sévères et chez certains hémizygotes dits atypiques, la maladie peut prendre une forme particulière et se limiter à des manifestations cliniques modérées ou limitées à l’atteinte d’un organe. On définit ainsi le variant cardiaque (cardiomyopathie hypertrophique ou remodelage ventriculaire gauche), le variant rénal (protéinurie, insuffisance rénale progressive) et le variant vasculaire (accident vasculaire cérébral inaugural).
Dans ce contexte, le dosage de l’activité enzymatique est un outil qui peut être utilisé pour le dépistage des hémizygotes atypiques et des hétérozygotes, sachant toutefois qu’une activité normale chez ces dernières n’exclut pas formellement l’absence de la maladie.
La prévalence estimée de la maladie de Fabry dans la population générale étant faible puisqu’elle se situe autour d’une naissance sur 100 000, une campagne de dépistage néonatal systématique n’aurait donc aucun sens. En revanche, les prévalences observées dans certaines populations à risque, présentant des signes cliniques compatibles avec les complications de la maladie de Fabry, sont beaucoup plus importantes. La première étude prospective, menée sur le variant cardiaque en 1995 (230 patients japonais présentant une hypertrophie ventriculaire gauche) a montré une prévalence de 3 % [2]. Deux autres études ont été réalisées sur des patients présentant une cardiomyopathie hypertrophique, l’une sur une population de 153 hommes [3] et l’autre de 34 femmes [4], montrant des prévalences de 3,9 % et de 12 % respectivement. La méthodologie employée dans la dernière étude est toutefois questionnable devant faire interpréter avec beaucoup de prudence ce chiffre de 12 %, très probablement surévalué. Des études sur de plus larges cohortes ont été menées sur le variant rénal dans des populations de patients hémodialysés chroniques. Les prévalences calculées dans des populations d’hommes de 514 patients japonais [5] et 508 hollandais [6] sont de 1,2 % et 0,22 % respectivement. Une étude autrichienne sur 2 480 hommes et femmes hémodialysés, représentant 80,1 % de la population des hémodialysés du pays permet de calculer une prévalence de 0,16 % (4 hommes sur 1 516 et 0 femme sur 964). Il appa raît donc que ces prévalences précédemment publiées à partir de registres d’hémodialysés européens et américains (données rétrospectives) et qui sont inférieures à 0,02 %, sont largement sous-estimées en absence de dépistage systématique. Bien que les prévalences dans ces populations à risque soient relativement faibles (comprises entre 0,16 et 3,9 %) et vraisemblablement plus proches de la limite inférieure de cet intervalle, le dépistage systématique dans ces populations se justifie par le fait qu’il peut non seulement permettre de poser un diagnostic pour le cas index mais aussi d’ouvrir des possibilités de prévention pour ses apparenté(e)s après recueil et construction de l’arbre généalogique. Un diagnostic précoce avant apparition des signes cliniques de la maladie, devient ainsi possible pour des sujets qui ne se savaient pas à risque. Ces programmes de dépistage de malades non diagnostiqués dans des populations à risque sont d’autant plus intéressants que la maladie de Fabry est l’une des rares maladies orphelines qui possède un traitement spécifique par substitution de l’enzyme déficiente [7].
L’α-galactosidase A est une enzyme lysosomale hydrolytique impliquée dans le catabolisme des glycosphingolipides. La Figure 1A illustre la réaction d’hydrolyse de l’a-galactose en position terminale du Gb3, substrat majoritairement accumulé dans la maladie de Fabry. Afin de réaliser les mesures de l’activité enzymatique de l’α-galactosidase A dans différentes matrices biologiques, les techniques existantes sont nombreuses. Le dosage de l’activité enzymatique consiste en la mesure du produit issu de la réaction enzymatique. Une première approche est fondée sur la quantification du galactose : en présence de l’enzyme fonctionnelle, le galactose libéré par le Gb3 subit une déshydrogénation par une galactose déshydrogénase en présence de NAD+. Lors de cette réaction, le NAD+ est réduit en NADH,H+, molécule dont l’absorbance à 340 nm est proportionnelle à la quantité de galactose. Toutefois, cette technique est peu sensible et peu spécifique. Une autre approche a consisté en un radio-marquage spécifique au tritium de la position 6 du galactose en position terminale du Gb3, l’activité enzymatique étant calculée par la mesure de la radioactivité du galactose libéré. Cette technique, relativement longue, est très peu utilisée en raison des contraintes de radio-protection qui lui sont associées. Les techniques les plus utilisées font appel à l’utilisation de substrats artificiels dont le produit de la réaction possède des propriétés spectrales exploitables en milieu biologique complexe. Citons le p-nitrophényl-a-galactose qui, en présence de l’enzyme fonctionnelle, libère une molécule de p-nitrophénol dont le maximum d’absorbance se situe à 405 nm. Toutefois le substrat artificiel le plus largement utilisé est le 4-méthyl umbelliféryl-a-galactose qui libère le 4-méthyl umbelliférone, molécule fluorescente (longueur d’onde d’excitation λex = 360 nm ; longueur d’onde d’émission λém = 446 nm) en milieu alcalin (Figure 1B). Les mesures de fluorescence présentent l’avantage d’être sensibles et spécifiques par leurs longueurs d’ondes d’excitation et d’émission. Par ailleurs, le risque d’interférences est réduit car peu de molécules sont fluorescentes dans les matrices biologiques. L’utilisation d’un substrat artificiel produisant une molécule fluorescente associé à un traitement de l’échantillon adapté (assurant une bonne conservation de l’enzyme et permettant de s’affranchir des interférents biologiques) apparaît donc comme la technique de choix pour la détermination des activités enzymatiques sur de grandes séries de prélèvements correspondant à des populations à risques. ‡
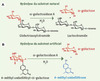 |
Figure 1. Hydrolyse d’un substrat naturel (A) et d’un substrat artificiel (B) de l’α-galactosidase A. |
Références
- Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961; 190 : 372–3. [Google Scholar]
- Nakao S, Takenada T, Maeda M, et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med 1995; 333 : 288–93. [Google Scholar]
- Sachdev B, Takenada T, Teraguchi H, et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002; 105 : 1407–11. [Google Scholar]
- Chimenti C, Pieroni M, Morgante E, et al. Prevalence of Fabry disease in female patients with late-onset hypertrophic cardiomyopathy. Circulation 2004; 110 : 1047–53. [Google Scholar]
- Nakao S, Kodama C, Takenaka T, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a renal variant phenotype. Kidney Int 2003; 64 : 801–7. [Google Scholar]
- Linthorst GE, Hollak CEM, Korevaar JC, et al. Alpha-galactosidase A deficiency in Dutch patients on dialysis : a critical appraisal of screening for Fabry disease. Nephrol Dial Transplant 2003; 18 : 581–4. [Google Scholar]
- Kotanko P, Kramar R, Devrnja D, et al. Results of a nationwide screening for Anderson-Fabry disease among dialysis patients. J Am Soc Nephrol 2004; 15 : 1323–9. [Google Scholar]
- Wilcox WR, Banikazemi M, Guffon N, et al. Long-term safty and efficacy on enzyme replacement therapy for Fabry disease. Am J Hum Genet 2004; 75 : 65–74. [Google Scholar]
Liste des figures
|
Figure 1. Hydrolyse d’un substrat naturel (A) et d’un substrat artificiel (B) de l’α-galactosidase A. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.