")
")
| Issue |
Med Sci (Paris)
Volume 21, Décembre 2005
Métabolisme glucidolipidique et risque cardiovasculaire: Nouvelle approches
|
|
|---|---|---|
| Page(s) | 40 - 42 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20052111s40 | |
| Published online | 15 November 2005 | |
L’angiokératome dans le diagnostic de la maladie de Fabry
Angiokeratomas and diagnostic of Fabry disease
Service de Dermatologie, Hôpital Fournier, CHU de Nancy, 36, quai de la Bataille, 54000 Nancy, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les différents aspects dermatologiques de la maladie de Fabry sont étudiés ainsi que les mécanismes pathogéniques qui en sont la cause. Les traitements sont discutés
Abstract
Angiokeratomas and other dermatologic aspects of Fabry disease are described. Their pathologic significance is analysed. The treatments are discussed
© 2005 médecine/sciences - Inserm / SRMS
La maladie de Fabry a été initialement décrite par deux dermatologues, Johannes Fabry [1] en 1898 sous la dénomination de purpura hemorrhagica nodularis, puis d’angiokeratoma corporis diffusum en 1915 et William Anderson [2], de façon indépendante mais également en 1898.
Il s’agit d’une des maladies génétiques les plus fréquentes rencontrées par le dermatologue et si son incidence est estimée à 1 pour 40 000 naissances masculines, une sous-estimation est probable. L’existence depuis quelques années d’un traitement enzymatique substitutif incite à la reconnaissance de cette maladie et à la mise en place de recommandations quant à l’utilisation de celui-ci [3, 4].
Les manifestations dermatologiques peuvent révéler la maladie par un simple examen clinique, et ceci montre tout l’intérêt de connaître la description de l’angiokératome [5].
Signes cliniques dermatologiques
Angiokératome
Il s’agit d’une dilatation vasculaire (papule télangiectasique) dont la surface est kératosique à la palpation, se vidant en grande partie lors de la vitro-pression. Les angiokératomes sont multiples, de petite taille, punctiformes, pseudopurpuriques et augmentent avec le temps pouvant constituer une éruption profuse, prédominant sur la racine des cuisses et sur les fesses (en caleçon) (Figure 1). Cependant, cette topographie, si elle est très évocatrice, n’est pas constante et d’autres atteintes sont également possibles : le tronc, les doigts, le scrotum, la muqueuse orale et les conjonctives. Ils apparaissent pendant l’enfance ou l’adolescence mais ne sont pas les premiers signes cliniques.
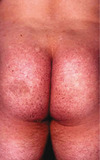 |
Figure 1. Angiokératomes des fesses. |
Dans une étude portant sur 366 patients [6], ils sont constatés chez 78 % des garçons et 50 % des filles. L’âge moyen d’apparition est de 17,9 ans chez le garçon et 29,1 ans chez la fille. À l’âge adulte, 10 % des hommes ayant la maladie de Fabry n’ont pas d’angiokératomes alors qu’on en observe chez 30 % des femmes hétérozygotes.
La biopsie cutanée d’un angiokératome dans le contexte de la maladie de Fabry permet de montrer, lors de l’étude histopathologique, la dilatation des capillaires de la papille dermique, l’allongement des crêtes épidermiques et une hyperkératose orthokératosique souvent moins marquée que dans les autres types d’angiokératomes. Il n’y a pas d’anomalie dermique ou hypodermique. La dilatation capillaire serait la conséquence d’une fragilité vasculaire par dépôts de globotriaosylcéramide dans les lysosomes des cellules endothéliales, bien mis en évidence en microscopie et immunomicroscopie électroniques [7].
Autres signes dermatologiques
L’hypohidrose est quasi constante chez l’adulte jeune, responsable d’une intolérance à l’effort physique et à la chaleur et parfois d’hyperthermie. L’anhidrose est plus rare. Ces anomalies sont dues à l’atteinte nerveuse mais également aux inclusions de glycosphingolipides dans les cellules sudorales eccrines [7, 8]. De rares cas d’hyperhidrose ont été rapportés.
D’autres signes ont également été mentionnés mais sont moins constants : cheveux fins, dépilation, érythème palmaire, acrocyanose, lymphœdème (mains, pieds, paupières), visage évoquant l’acromégalie.
Diagnostic différentiel
Le diagnostic d’angiokératome sur la clinique et l’histopathologie n’est pas suffisant pour affirmer une maladie de Fabry, de nombreuses affections pouvant comporter comme signes cliniques des angiokératomes.
Si les angiokératomes sont diffus, plusieurs maladies métaboliques peuvent être évoquées [9] : fucosidose, α-galactosidose, maladie de Kanzani, α-mannosidose, aspartylglucosaminurie, galactosialidose. Des angiokératomes ont également été occasionnellement rapportés dans le syndrome de Turner, la sclérose tubéreuse de Bourneville et la dermatomyosite. Il faut également signaler des observations d’angiomes caverneux familiaux avec manifestations cutanées évocatrices d’angiokératomes [10] et des cas sporadiques.
Les angiokératomes de topographie locale doivent faire envisager d’autres diagnostics [11] :
L’angiokératome solitaire, tumoral (Figure 2) : nodulaire, parfois multiple, il est ubiquitaire et souvent acquis après un traumatisme. S’il est le siège d’une thrombose, son aspect noir peut simuler un mélanome. Il doit donc être distingué d’un mélanome, d’une maladie de Kaposi, d’un angiome verruqueux (composante vasculaire profonde). Le traitement en est l’excision.
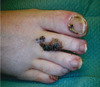
Figure 2. Angiokératome solitaire.
Les angiokératomes du scrotum et de la vulve décrits par Fordyce en 1896 [12] sont constatés fréquemment à partir de 40 ans (Figure 3). De 1 à 5 mm de diamètre, il s’agit de petites papules multiples, kératosiques, violacées, voire noires, « grains de caviar », distribuées sur le scrotum, la verge, la vulve, parfois le pubis et la partie haute des cuisses. Ils sont probablement la conséquence d’une hyperpression veineuse et aucune anomalie métabolique n’est constatée.
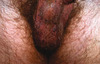
Figure 3. Angiokératomes de Fordyce du scrotum.
Les angiokératomes décrits par Mibelli en 1869 [11] sont rares, touchant plus souvent les extrémités (face dorso-latérale des doigts ou des orteils), plus rarement le dos des mains et des pieds que les coudes, les genoux ou les fesses. Ils sont plus fréquents chez la fille, associés à une acrocyanose, et souvent familiaux.
Le syndrome APACHE (acral pseudolymphomatous angiokeratoma of children) est à tort classé dans les angiokératomes des extrémités alors que, si l’aspect clinique peut évoquer des angiokératomes, l’étude histopathologique fait discuter un pseudo-lymphome voire une hyperplasie angio-lymphoïde [13].
L’angiokératome circonscrit næviforme de Fabry (II) ou angiome serpigineux de Hutchinson (1889) [14] est une malformation congénitale rare caractérisée par des angiokératomes de 1 à 5 mm groupés en une ou plusieurs plaques sur un segment de membre, sur une fesse, avec parfois une disposition linéaire ou métamérique. Ils deviennent plus kératosiques avec l’âge et saignent fréquemment. Isolés ou associés à une malformation vasculaire sous-jacente, ils nécessitent des explorations vasculaires. L’étude histopathologique cutanée montre des ectasies capillaires avec atteinte plus profonde dermique ou hypodermique.
D’autres anomalies vasculaires peuvent être confondues avec des angiokératomes :
l’angiome stellaire et les taches rubis correspondent à une dilatation des vaisseaux dermiques superficiels, plus ou moins linéaire, disparaissant à la vitropression. Ils sont fréquents, tout particulièrement chez l’enfant et la femme enceinte et lors de maladies telles que la cirrhose, la sclérodermie, la radiodermite, la rosacée… ;
les télangiectasies linéaires unilatérales nævoïdes apparaissent progressivement à la fin de l’enfance ou pendant l’adolescence : il s’agit d’un lacis de télangiectasies disséminé sur un territoire loco-régional voire métamérique. Elles ne s’accompagnent d’aucune autre anomalie ;
les télangiectasies essentielles progressives constituent d’innombrables chevelus capillaires d’extension progressive, sans topographie particulière ;
enfin, des télangiectasies sont fréquemment constatées chez des patients souffrant de maladies systémiques telles que la sclérodermie, la maladie de Rendu-Osler, la mastocytose… ;
les autres malformations vasculaires, dont les lymphangiectasies et le lymphangiome, sont rarement confondues avec des angiokératomes.
Traitement
Le traitement des angiokératomes n’est pas indispensable et il n’est envisagé qu’à la demande du patient. Différents traitements ont été utilisés dont la cryothérapie, l’électrocoagulation et la photovolatilisation au laser CO2. Plus récemment, les lasers vasculaires, tels que le laser KTP ont été proposés [15].
Conclusion
Il est donc important de connaître cette petite tumeur vasculaire qu’est l’angiokératome. Dans la maladie de Fabry, le nombre ou la précocité d’apparition de ceux-ci ne sont pas un critère de gravité ou d’évolutivité imposant un traitement par a-galactosidase A recombinante. De même, il n’y a pas pour l’instant d’étude montrant une régression lors du traitement substitutif [16, 17].
Références
- Fabry J. Ein Beitrag zur Kenntnis der Purpura haemorrhagica nodularis (Purpura papulosa hemorrhagica Hebrae). Arch Dermatol Syphilol 1898; 43 : 187–200. [Google Scholar]
- Anderson W. A case of Angeio-keratoma. Br J Dermatol 1898; 10 : 113–7. [Google Scholar]
- Desnick RJ, Brady R, Barranger J, et al. Fabry disease, an under-recognized multisystemic disorder : expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med 2003; 138 : 338–46. [Google Scholar]
- Germain DP. La maladie de Fabry. Aspects cliniques et génétiques. Perspectives thérapeutiques. Rev Med Interne 2000; 21 : 1086–103. [Google Scholar]
- Mahé E, Hadj-Rabia S, Chauveau D, et al. Maladie de Fabry. Place du dermatologue et progrès thérapeutique. Ann Dermatol Venereol 2005; 132 : 171–6. [Google Scholar]
- Mehta A, Ricci R, Widmer U, et al. Fabry disease defined : baseline clinical manifestation of 366 patients in the Fabry outcome survey. Eur J Clin Invest 2004; 34 : 236–42. [Google Scholar]
- Kanekura T, Fukushige T, Tsuyama S, et al. Immunoelectron-microscopic detection of globotriaosylceramide accumulated in the skin of patients with Fabry disease. Br J Dermatol 2005; 153 : 544–8. [Google Scholar]
- Kang WH, Chun SI, Lee S. Generalized anhidrosis associated with Fabry’s disease. J Am Acad Dermatol 1987; 17 : 883–7. [Google Scholar]
- Kanitakis J, Allombert C, Doebelin B, et al. Fucosidosis with angiokératome. Immunohistochemical and electronmicroscopic study of a new case and literature review. J Cutan Pathol 2005; 32 : 506–11. [Google Scholar]
- Linthorst GE, De Rie MA, Tjiam KH, et al Misdiagnosis of Fabry disease : importance of biochemical confirmation of clinical or pathological suspicion. Br J Dermatol 2004; 150 : 575–7. [Google Scholar]
- Masouyé I, Saurat JH. Angiokératomes. In : Dermatologie et infections sexuellement transmissibles, 4e ed. Paris : Masson, 2004 : 723–5. [Google Scholar]
- Fordyce JA. Angiokeratoma of the scrotum. J Cutan Dis 1896; 14 : 81–9. [Google Scholar]
- Kim Y, Dawes-Higgs E, Mann S, et al. Acral pseudolymphomatous angiokeratoma of children (APACHE). Australas J Dermatol 2005; 46 : 177–80. [Google Scholar]
- Degos R. Angiokératose circonscrite næviforme (Fabry II). Angiome serpigineux (Hutchinson). In : Dermatologie. Paris : Flammarion Médecine-Sciences, 1980 : 774 p. [Google Scholar]
- Gorse SJ, James W, Murison MSC. Successful treatment of angiokeratoma with potassium tritanyl phosphate laser. Br J Dermatol 2004; 150 : 620–2. [Google Scholar]
- Möhrenschlager M, Henkel V, Ring J. Fabry disease. More than angiokeratomes. Arch Dermatol 2004; 140 : 1526–8. [Google Scholar]
- Ries M, Schiffmann R. Fabry disease : angiokeratoma, biomarker and the effect of enzyme replacement therapy on kidney function. Arch Dermatol 2005; 141 : 904–5. [Google Scholar]
Liste des figures
|
Figure 1. Angiokératomes des fesses. |
| Dans le texte | |
|
Figure 2. Angiokératome solitaire. |
| Dans le texte | |
|
Figure 3. Angiokératomes de Fordyce du scrotum. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.