")
")
| Issue |
Med Sci (Paris)
Volume 18, Number 11, Novembre 2002
|
|
|---|---|---|
| Page(s) | 1146 - 1154 | |
| Section | Repères : Lexique | |
| DOI | https://doi.org/10.1051/medsci/200218111146 | |
| Published online | 15 novembre 2002 | |
Phylogénie et évolution moléculaires
Bio-informatique (5)
Molecular phylogeny and evolution
Phylogénie, Bio-informatique et Génome, Cnrs UMR 7622, Université Pierre et Marie Curie, 9, quai Saint-Bernard, Bâtiment C, 75005 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La phylogénie moléculaire a pour but de reconstruire les relations de parenté entre des séquences de nucléotides ou d’acides aminés. On peut ainsi étudier les relations de parenté entre les espèces qui les portent mais, aussi, l’évolution du génome. En particulier, pour chaque famille multigénique, on peut déterminer l’importance relative des événements de duplications et de transferts horizontaux de gènes. La fiabilité des méthodes de reconstruction phylogénétique repose sur la compréhension des mécanismes d’évolution des séquences, un domaine qui a beaucoup progressé ces dernières années. Cela a abouti à une vision sans cesse plus correcte de l’arbre universel du vivant. L’étude des contraintes fonctionnelles agissant sur les protéines bénéficie de ces avancées. En particulier, la détection, dans une protéine, des positions qui sont soumises à une sélection darwinienne est devenue assez performante, permettant de prédire les substitutions à l’origine d’un changement de fonction et donc de guider les études expérimentales.
Abstract
The aim of molecular phylogenetics is to reconstruct the genealogical relationships between nucleic or amino acid sequences. The phylogeny of the species bearing these sequences can be then inferred, but also the molecular evolution of the genomes can be analysed. For gene families, the relative importance of gene duplications and horizontal gene transfers can be examined. The reliability of the methods used to infer molecular phylogenies relies on the accuracy of our knowledge about the mechanisms of sequence evolution. Tremendous progresses have been done in this field of research in the last few years that helped us to get a better picture of the universal tree of life. The methods to analyse the selective constraints acting at the protein level have also been much improved. In particular, the detection of the sites in a protein which are under positive darwinian selection is getting more powerful. It is now possible to use the prediction about the critical residues associated to functional shift as a guide for further experimental approaches.
© 2002 médecine/sciences - Inserm / SRMS
Comme l’a si bien exprimé Dobzhansky (1900-1975), « rien n’a de sens en biologie, si ce n’est à la lumière de l’évolution ». À l’heure où la génomique et la « postgénomique » produisent d’importantes quantités de données expérimentales, un des facteurs limitant reste leur analyse bio-informatique. Les approches évolutives de comparaison de séquences de nucléotides et/ou d’acides aminés, apparues il y a une trentaine d’années, constituent un outil de choix. Nous allons tout d’abord nous intéresser, ici, à la phylogénie moléculaire, qui permet de retracer les relations généalogiques entre les gènes (et donc entre les espèces qui les portent), puis aux méthodes qui permettent de détecter, au niveau nucléotidique, l’action de la sélection naturelle.
Principes de reconstruction des phylogénies moléculaires
Pour construire une phylogénie, il faut disposer de caractères comparables entre tous les objets (c’est-à-dire gènes ou espèces) que l’on veut analyser. En d’autres termes, les objets analysés doivent être « suffisamment similaires » pour être comparés. Si c’est le cas, on dit de ces caractères qu’ils sont homologues c’est-à-dire qu’on formule l’hypothèse selon laquelle la similitude observée est due au fait que les caractères sont issus d’un ancêtre commun et qu’ils se sont progressivement modifiés au fil des générations. Pour les séquences de protéines ou d’ADN, cette étape de comparaison est celle de l’alignement. Les programmes d’alignement automatique sont très efficaces pour les régions de forte similitude (plus de 50 % des positions de l’alignement portent des nucléotides ou des acides aminés identiques) mais ne le sont pas pour les séquences plus divergentes, même si l’utilisation de la structure tridimensionnelle améliore parfois sensiblement les résultats. Après un alignement automatique, une étape cruciale, trop souvent négligée par les non spécialistes, est donc de l’affiner manuellement et, avant tout, de retirer de l’analyse les régions où l’alignement est « ambigu » (choix souvent subjectif, car la mise au point de méthodes automatiques se révèle extrêmement délicate) [1].
Une fois que l’on dispose d’un alignement non ambigu (c’est-à-dire où toutes les positions sont homologues), il faut trouver, parmi tous les arbres phylogénétiques possibles, celui qui correspond à l’histoire évolutive de toutes ces séquences. Malheureusement, le nombre d’arbres possibles augmente de manière exponentielle avec le nombre d’espèces analysées. Ainsi, pour 50 espèces, il existe 2,8.1074 arbres possibles. Un ordinateur extrêmement performant qui analyserait un milliard d’arbres par seconde aurait ainsi besoin de 1058 années pour mener à bien le calcul exhaustif de tous les arbres. Dans la pratique, il est donc hors de question de parcourir tout l’espace de recherche et il faut donc accepter que le programme ne produise pas nécessairement le meilleur arbre mais, au moins, un arbre s’en approchant. On dit de tels programmes qu’ils sont des heuristiques et on peut citer comme exemple de ces algorithmes le réarrangement des branches dans l’arbre ou la redistribution aléatoire de l’ordre d’agglomération (voir glossaire) des espèces. En pratique, on est obligé de faire un compromis entre temps de calcul et efficacité de la recherche, ce qui fait que l’on est rarement certain d’avoir trouvé le meilleur arbre, même si l’on est toujours sûr d’avoir trouvé un très bon arbre (c’est-à-dire proche du meilleur).
La question la plus intéressante est de savoir trouver, à partir des séquences actuelles, quel est le « vrai » arbre phylogénétique. Toutes les méthodes de reconstructions sont fondées sur un critère quantitatif et recherchent l’arbre qui minimise (ou maximise) ce critère. Avant de détailler ce critère, voyons en quoi consiste l’information apportée par l’alignement des séquences. Il peut s’agir d’une mutation, changeant par exemple le nucléotide A en C (se traduisant par le changement de l’acide aminé lysine en tyrosine à la position i). Si cette mutation s’est fixée dans l’ancêtre commun de certaines espèces (Figure 1), on peut envisager le regroupement de ces espèces à l’exclusion de toutes les autres, car ce sont les seules à posséder un Y (tyrosine) à la position i. Malheureusement, une mutation peut aussi aisément créer du « bruit », en particulier en raison des phénomènes de réversion et de conversion (Figure 1). Ainsi, l’existence de substitutions multiples à la même position explique bien la difficulté des reconstructions des phylogénies moléculaires: plus il y a de signal (c’est-à-dire de mutations), plus il y a de bruit (c’est-à-dire de convergences et de réversions). L’idéal serait d’avoir une infinité de positions évoluant lentement (donc peu bruitées) mais, dans la réalité, nous sommes en présence d’une quantité limitée de positions évoluant à des vitesses très variables.
 |
Figure 1. Information et bruit lors de la construction des phylogénies moléculaires. Sur cet arbre phylogénétique est représentée l’évolution de trois positions, i, j et k d’une protéine. La position i n’a subi qu’une seule substitution, se traduisant par le changement de l’acide aminé lysine (K) en tyrosine (Y). Si cette mutation s’est fixée dans l’ancêtre commun des espèces 9-15, on peut envisager le regroupement des espèces 9-15 à l’exclusion de toutes les autres, car ces espèces sont les seules à posséder un Y à la position i. Dans ce cas, le signal apporté par la position i est peu bruité. Malheureusement, une mutation peut aussi aisément créer du « bruit », ce qui peut amener à des artéfacts de reconstruction: c’est le cas des positions j et k où deux substitutions ont eu lieu, une réversion en j, et une convergence en k. Imaginons par exemple que l’ancêtre commun des espèces 22-27 fixe une mutation qui change la valine (V) en isoleucine (I) à la position j, mais que l’ancêtre de l’espèce 27 fixe une mutation qui change I en V (ce que l’on appelle une réversion), alors l’espèce 27 va être illégitimement exclue du groupe 22-27. De même, si les ancêtres communs des groupes 4-6 et 16-21 fixent, de façon indépendante et à cette même position k, une mutation qui change l’acide aspartique (D) en acide glutamique (E) (ce que l’on appelle une convergence), alors les espèces 4-6 seraient indûment groupées avec les espèces 16-21. |
Dans un précédent article (→), nous avons détaillé deux approches, proposées dès les années 1960: le maximum de parcimonie et les méthodes de distances (voir glossaire). Toutes deux ont l’avantage d’être très rapides, mais se sont révélées moins performantes que le maximum de vraisemblance [2]. Schématiquement, cette méthode estime la probabilité d’observer les données (ici l’alignement) sous une hypothèse donnée (ici l’arbre phylogénétique), ce que l’on appelle vraisemblance. On choisit, comme étant le meilleur, l’arbre (c’est-à-dire l’hypothèse) qui maximise le critère de vraisemblance. Pour calculer la vraisemblance, d’autres informations sont nécessaires comme la probabilité de changement d’un nucléotide vers un autre par exemple transition versus transversion (voir glossaire) ou la longueur des branches de l’arbre. On appelle modèle d’évolution des séquences l’ensemble de ces hypothèses portant sur les processus d’évolution. Il faut néanmoins noter que toutes les informations du modèle n’ont pas nécessairement besoin d’être spécifiées (en particulier la longueur des branches), car elles peuvent être estimées à partir de l’alignement et de l’arbre en maximisant la vraisemblance. Des études empiriques et par simulation ont montré que les méthodes de maximum de vraisemblance sont actuellement les plus efficaces pour retrouver le véritable arbre phylogénétique. Le facteur limitant important de ces méthodes est le temps de calcul: il faut par exemple plusieurs heures de calcul pour estimer seulement la vraisemblance d’un arbre ainsi que les paramètres (par exemple les longueurs de branches), pour une centaine de séquences.
(→) m/s 1995, n° 8, p. I-XIII
Récemment, les méthodes de maximum de vraisemblance ont énormément progressé grâce à l’amélioration des modèles d’évolution des séquences. En effet, plus les hypothèses du modèle sont proches de la manière dont les séquences ont évolué, plus la fiabilité du maximum de vraisemblance sera grande. En particulier, les premiers modèles faisaient l’hypothèse que tous les sites évoluaient à la même vitesse. Or, comme les contraintes fonctionnelles portant sur les différentes positions d’une protéine sont différentes, certaines positions vont évoluer très lentement (voire être invariables, comme les acides aminés impliqués dans un site actif) ou, au contraire, très vite. Les modèles actuellement utilisés représentent cette variabilité au moyen d’une distribution gamma (voir glossaire), ce qui améliore souvent les inférences. Par exemple, les microsporidies, parasites intracellulaires de nombreux métazoaires, étaient considérés comme des eucaryotes d’émergence très précoce sur la base de la comparaison de leur ARN ribosomique [3]. La prise en compte de l’hétérogénéité de la vitesse d’évolution des positions a radicalement changé ces conclusions, en les plaçant, en réalité, au milieu des champignons, ce qui a été confirmé par d’autres informations [4].
La phylogénie à l’heure du génome
Les progrès du séquençage ont amené un flot de données qui se prêtent particulièrement bien à l’analyse phylogénétique. L’utilisation simultanée de nombreux gènes, permettant d’obtenir plusieurs milliers de positions homologues, a permis de résoudre plusieurs questions débattues de longue date. À grande échelle évolutive, on peut citer en exemple la démonstration de la monophylie des algues rouges et des algues/plantes vertes [5], qui confirme définitivement l’origine unique des chloroplastes par endosymbiose d’une cyanobactérie. À l’intérieur des plantes vertes, les gnétales ont longtemps été considérées comme proche des angiospermes (plantes à fleurs) à cause de plusieurs caractères communs comme la double fécondation, alors qu’en fait ce sont des conifères [6] qui ont acquis les caractères précédents de manière convergente, montrant que le problème des substitutions multiples évoqué plus haut pour les données moléculaires affecte aussi les données morphologiques. Enfin, la question épineuse des relations de parenté entre les différents ordres de mammifères a très sérieusement progressé. En particulier, la monophylie des rongeurs, que les premières études de phylogénie moléculaire rejetaient [7, 8], a finalement été confirmée [9, 10], ce qui a clos un long conflit avec les anatomistes et les paléontologistes, convaincus, à juste titre, de leur monophylie. Cela a aussi validé l’hypothèse selon laquelle la non-monophylie apparente des rongeurs était le résultat d’un artéfact de reconstruction, connu sous le nom d’attraction des longues branches (voir glossaire) [11] (→). Ces études ont aussi confirmé la proche parenté de ce groupe avec les lagomorphes (lapin), mais ont étonnamment montré une proche parenté avec les primates à l’exclusion des carnivores, des cétacés, des chauve-souris ou des artiodactyles (vache) [9, 10]. Ainsi, la souris - l’organisme modèle par excellence - est plus proche de l’homme qu’on ne le pensait, ce qui augmente l’intérêt de l’étude de son génome pour aider à décrypter le génome humain. (→→) Outre leurs applications purement taxonomiques, les données de séquençage des génomes ont ouvert la voie à de multiples recherches en évolution moléculaire. De nombreux gènes sont en effet présents en de multiples copies à l’intérieur d’un même organisme, et leur phylogénie permet de mieux comprendre leur évolution. Prenons l’exemple du gène codant pour le récepteur de la dopamine D1, gène qui a subi plusieurs duplications au cours de l’histoire des vertébrés (Figure 2). L’analyse de nombreux gènes [12] suggère que deux duplications du génome ont eu lieu tôt dans l’histoire des vertébrés, ce qui est en accord avec la célèbre hypothèse 2R d’Ohno [13]. Cette théorie, fondée essentiellement sur des considérations de taille des génomes, propose qu’il y ait eu deux événements de tétraploïdisation chez un ancêtre des vertébrés et que la diversification des vertébrés ait été facilitée par ce stock de gènes redondants, qui ont pu acquérir de nouvelles fonctions (puisqu’il suffit qu’une seule copie garde la fonction ancestrale). L’étude du génome de la plante Arabidopsis thaliana sug-gère que le scénario pourrait être beaucoup plus compliqué. En effet, la moitié des gènes récemment dupliqués l’ont été en tandem (c’est-à-dire par crossing over inégal) et l’autre moitié via la duplication de fragments de chromosomes [14]. L’explication la plus communément admise est qu’une tétraploïdisation a eu lieu il y a environ 110 millions d’années et que seuls certains fragments de chromosomes ont été conservés en double exemplaire. De même, chez les vertébrés, les duplications en tandem (par exemple, dans la famille des hémoglobines) constituent un phénomène récurrent [15]. Une alternative à l’hypothèse 2R propose simplement qu’il y ait eu de nombreuses duplications soit en tandem soit de fragments de chromosomes au cours de la diversification précoce des vertébrés [16]. Le choix entre ces deux hypothèses est difficile à faire, bien qu’elles produisent des prédictions très différentes sur les phylogénies de gènes [15]. En effet, les phylogénies fondées sur un seul gène sont souvent mal résolues (trop peu de signal) et sont rendues difficiles par les changements de vitesse d’évolution (d’où des artéfacts dus à l’attraction des longues branches) (Figure 2).
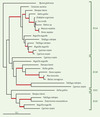 |
Figure 2. Phylogénie du gène D1 codant pour un récepteur de la dopamine. Cet arbre a été « raciné » sur la myxine (un poisson dépourvu de mâchoires), c’est-à-dire que l’on a décidé, sur la base d’informations extérieures (ici paléontologiques et morphologiques) de placer le début de l’histoire de ces séquences entre la myxine et les gnathostomes (poissons à mâchoires). En effet, à partir des seules données moléculaires, il est très difficile de positionner le début de l’histoire (à moins de faire de nombreuses hypothèses). Le gène D1 s’est dupliqué en quatre copies chez l’ancêtre des vertébrés (notées ici A, B, C et D), qu’on appelle des paralogues (voir glossaire). De plus, le paralogue A s’est luimême dupliqué au sein des poissons, ce qui prouve que de tels événements sont fréquents. La résolution de l’arbre n’est pas bonne (les branches significativement robustes sont représentées en rouge), sans doute à cause du petit nombre de positions (~ 300). On notera que la phylogénie n’est pas parfaitement conforme à celle des espèces, comme le montre la non-monophylie des poissons pour D1A et D1B. Un scénario possible pour l’évolution de ce récepteur est que les quatre copies ont été engendrées par les deux événements hypothétiques de tétraploïdisation, que certaines ont été perdues dans plusieurs lignées de vertébrés et que d’autres événements de duplication (en tandem ou par tétraploïdisation) ont eu lieu chez les poissons téléostéens. En particulier, on notera l’absence des gènes D1C et D1D chez les mammifères, qui n’est pas due à un problème d’échantillonnage mais à deux pertes. Pour ce gène impliqué dans le système nerveux central, contrairement aux idées préconçues, les poissons ont plus de gènes que les mammifères. La barre correspond à un nombre moyen de 0,1 changement par position. |
(→) m/s1995, n° 8, p. I-XIII
(→→) m/s2000, n° 1, p. 31
L’étude des transferts horizontaux de gènes est un autre champ d’application des phylogénies de gènes. L’échange de matériel génétique entre espèces différentes est connu depuis longtemps, en particulier chez les procaryotes via les plasmides et les bactériophages. La résistance aux antibiotiques, en particulier, s’est propagée principalement de cette façon. L’analyse des génomes complets a montré que les transferts horizontaux de gènes étaient beaucoup plus fréquents qu’on ne le supposait [17]. Environ 30 % des gènes ne sont pas partagés entre deux souches d’Escherichia coli [18]. Si la façon la plus sûre de détecter le transfert d’un gène est de démontrer que la phylogénie de ce gène est significativement différente de la phylogénie des espèces, cette approche reste cependant lourde à mettre en œuvre. Les chercheurs utilisent des méthodes alternatives comme la recherche d’une composition anormale en nucléotides [19] ou d’une trop forte similitude avec un organisme éloigné [20]. Ce type d’approche a permis de montrer l’importance quantitative des transferts horizontaux de gènes [21]: on estime ainsi qu’environ 20 % des gènes de la bactérie hyperthermophile Thermotoga proviendraient des archaebactéries. Néanmoins, ces méthodes demeurent peu performantes [22] comme l’illustre l’analyse du génome humain, pour lequel il a été suggéré que 223 gènes humains auraient une origine procaryote récente, impliquant que les transferts horizontaux de gènes auraient eu lieu durant l’histoire des vertébrés [23]. Des analyses phylogénétiques détaillées ont toutefois montré que la plupart des transferts horizontaux de gènes avaient eu lieu beaucoup plus tôt dans l’évolution [24] et qu’en fait très peu, voire aucun, des gènes humains n’avaient une origine procaryote récente. Il est maintenant clair que les transferts horizontaux de gènes sont un facteur majeur dans l’adaptation des organismes, en particulier procaryotes, car ils constituent une source importante d’innovation [17]. Cependant, ils posent un problème considérable à la reconstruction de la phylogénie des espèces, puisqu’un échange trop important de patrimoine génétique finit par compromettre la notion même de patrimoine et, par conséquent, d’espèce. Certains auteurs ont même proposé qu’il n’existait pas de phylogénie des procaryotes [25]. On pourrait penser que les gènes les plus importants, impliqués dans de nombreuses interactions, ne peuvent pas se transférer, et constituent un « noyau » de gènes permettant de reconstruire la phylogénie des espèces (complexity hypothesis, [26]). Même si des transferts horizontaux de gènes ont été montrés pour certains de ces gènes, comme celui codant pour la protéine ribosomique rps14 [27], nous avons montré que cette hypothèse était vérifiée pour au moins une cinquantaine de gènes, aussi bien chez les bactéries que chez les archaébactéries [28, 29], et que donc la phylogénie des procaryotes existait et pouvait être reconstruite.
Évolution moléculaire
Malheureusement, les modèles d’évolution communément utilisés sont loin d’être parfaitement réalistes. Nous avons par exemple montré que, pour le cytochrome b, une protéine mitochondriale impliquée dans la respiration, la vitesse d’évolution de toutes les positions variables change au cours de l’évolution des vertébrés. De plus, ces changements ne semblent pas corrélés entre positions. Ce phénomène est appelé hétérotachie [30]. D’un point de vue biologique, ce comportement n’a rien de surprenant, puisqu’il paraît raisonnable de penser que les contraintes fonctionnelles des sites d’une protéine (et donc leurs vitesses d’évolution) vont changer au cours de l’évolution, et de façon indépendante selon les lignées. Or, tous les modèles d’évolution, comme ceux employant la distribution gamma, font des hypothèses beaucoup plus restrictives et supposent que la vitesse d’évolution d’une position donnée reste la même tout au long de l’histoire, même si elle peut varier entre positions. L’emploi de modèles simples est bien sûr dicté par des impératifs de temps de calcul et de complexité des algorithmes de reconstruction. Cependant, ces simplifications du modèle peuvent entraîner de graves erreurs de reconstructions [31]. Il est donc essentiel de développer des modèles qui reflètent autant que possible les observations réelles.
D’autres simplifications excessives ont été observées. Cela est en particulier le cas pour la modélisation des changements d’acides aminés où il est abusif d’utiliser la même matrice de substitutions pour tous les sites d’une protéine. Ces matrices, comme celle calculée par Margaret Dayhoff [32], ne représentent qu’une moyenne et ne reflètent jamais la probabilité de changement d’un site donné. Il est sans doute beaucoup plus raisonnable de proposer plusieurs matrices pour les différents types de positions (hélice α, feuillet β, extérieur versus intérieur de la protéine, etc.), comme cela commence à être fait [33]. En conclusion, même si notre compréhension de l’évolution des séquences est encore trop fragmentaire pour proposer le meilleur compromis entre simplification (nécessaire à la modélisation) et complexité (reflétant la réalité biologique), de nombreux problèmes sont identifiés et font à l’heure actuelle l’objet d’études de la part des phylogénéticiens.
Détection de la sélection au niveau moléculaire
Ainsi que nous l’avons vu, l’accumulation de différences au cours du temps dans des gènes homologues appartenant à des lignées évolutives indépendantes permet de reconstruire des phylogénies. Ces différences apparaissent d’abord sous la forme d’une mutation chez un individu, mais doivent ensuite envahir la population pour devenir observables. Ce phénomène est appelé fixation. La probabilité de fixation d’une mutation dépend de son impact sur le phénotype (favorable, nul ou défavorable) et donc de deux facteurs, la dérive génétique et la sélection naturelle. La dérive génétique est la variation aléatoire, au cours du temps, des fréquences des allèles dans des populations de taille finie. Même en l’absence de sélection (allèles neutres), un allèle finit toujours par envahir la population ou disparaître de celle-ci. Par ailleurs, si une mutation a des effets délétères, la probabilité qu’elle se fixe dans une population est réduite, c’est la sélection négative (purifying selection). Inversement, si une mutation favorise les individus qui la présentent, la probabilité qu’elle se fixe dans une population en est augmentée, c’est la sélection positive (Darwinian selection ou adaptive selection). Un enjeu fondamental de l’étude de l’évolution moléculaire est de mettre en évidence l’importance relative de la dérive génétique et de la sélection dans l’évolution des gènes. La théorie neutraliste de l’évolution moléculaire [34] suppose que la dérive génétique et la sélection négative sont les deux moteurs principaux de l’évolution des gènes, la sélection positive étant considérée comme très rare et épisodique. Cette vision de l’évolution implique que les séquences actuelles des gènes correspondent à un optimum constitué de deux fractions de sites nucléotidiques, plus ou moins importantes selon les gènes. Une première fraction est constituée des sites optimisés, tout changement étant délétère et éliminé par la sélection car il y a de fortes contraintes pour maintenir l’état observé du caractère. La seconde fraction est constituée des sites où tout changement est neutre (peu ou pas de contraintes sur l’état du caractère). Les événements de sélection adaptative, même s’ils sont rares, doivent être recherchés, car ils impliquent des changements de fonctions.
Différentes méthodes statistiques permettant d’identifier la sélection au niveau moléculaire ont été proposées. D’abord fondée sur la comparaison de deux séquences homologues [35], leur puissance a été augmentée en considérant un plus grand nombre de séquences et en tenant compte des relations phylogénétiques [36, 37]. Toutes ces méthodes reposent sur la classification en deux catégories des substitutions observées (Figure 3): (1) les mutations synonymes (silencieuses) qui modifient un codon, mais ne changent pas l’acide aminé codé, et (2) les mutations non-synonymes qui changent l’acide aminé. On peut estimer le nombre de substitutions synonymes par site synonyme (Ks) et le nombre de substitutions non-synonymes par site non-synonyme (Kns) qui se sont accumulées pendant le temps séparant deux séquences de leur ancêtre commun. Par ailleurs, on peut formuler l’hypothèse selon laquelle les mutations synonymes sont peu ou pas soumises à sélection (neutres) car elles ne modifient pas la protéine codée par un gène. Dans ce cas, le taux de substitution synonyme est un bon estimateur du taux de mutation [34]. Le taux de substitution non-synonyme dépend du niveau de contrainte sur une protéine. Trois situations peuvent alors se présenter.
Kns < Ks: le taux de substitution est inférieur au taux de mutation, ce qui indique l’existence d’une sélection négative éliminant des mutations non-synonymes. Plus Kns est faible par rapport à Ks, plus le nombre de substitutions non synonymes acceptables est faible.
Kns = Ks: il y a peu de contraintes sur la nature des acides aminés de la protéine.
Kns > Ks: les substitutions non-synonymes sont favorisées.
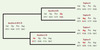 |
Figure 3. Estimation de la sélection au niveau moléculaire. Dans cet exemple simplifié, on considère quatre espèces actuelles (A, B, C, D) et quatre codons. Après avoir inféré les relations phylogénétiques entre ces espèces, on infère les séquences chez les trois ancêtres communs. Ensuite, pour chaque codon, on compte le nombre de substitutions synonymes (s), c’est-à-dire qui ne change pas l’acide aminé (indiqué en vert) et non-synonymes (ns), c’est-à-dire qui change l’acide aminé (indiqué en rouge). Dans le cas du premier codon, il y a eu trois mutations synonymes (s = 3) et une mutation non-synonyme (ns = 1). Comme on s’intéresse à la fréquence des changements, il faut corriger par le nombre potentiel de mutations synonymes (Ns) et non-synonymes (Nns). Par exemple, pour la valine (codé par GTN), il y a trois mutations synonymes possibles et six non-synonymes, alors que, pour le tryptophane (codé uniquement par UGG), il n’y a aucune mutation synonyme. Grâce à des méthodes plus ou moins sophistiquées, on calcule Ks (s/Ns) et Kns (ns / Nns). Ks est un estimateur du taux de mutation, Kns est un estimateur du taux de changement des acides aminés. Pour le premier codon, Kns est inférieur à Ks, ce qui indique une sélection négative qui empêche la fixation de certains changements d’acides aminés. Pour le deuxième codon, Kns est supérieur à Ks, ce qui indique une sélection positive qui favorise des changements d’acides aminés. |
Le facteur Kns est généralement très inférieur à Ks (cas 1), ce qui indique que la plupart des protéines sont globalement soumises à une forte sélection négative. Beaucoup plus rarement, Kns est supérieur à Ks et, dans ce cas, la sélection positive concerne essentiellement des protéines impliquées dans les interactions du type hôte-parasite ou gamètes mâle-femelle. Par exemple, chez l’homme, on observe que Kns est supérieur à Ks pour les gènes codant pour des immunoglobulines VH, des protéines du CMH et des ribonucléases. Le même phénomène est observé pour des protéines de virus comme celles de l’enveloppe du virus HIV ou d’hémagglutinine du virus influenza A (pour une liste plus complète, voir [36]). Une étude récente montre qu’un certain nombre de gènes impliqués essentiellement dans la fonction de reproduction chez l’homme accumulent préférentiellement les mutations non-synonymes [38]. La fixation à un rythme élevé de mutations changeant la nature des acides aminés correspondrait, dans les cas cités ci-dessus, à une adaptation permanente aux modifications des protéines du partenaire [36]. Les mesures du Kns et du Ks moyen pour un gène donné ne permettent pas d’identifier certains cas d’adaptation au niveau moléculaire, en particulier si le nombre de sites impliqués est faible. Dans ces cas, la mesure de la sélection position par position est alors plus informative, car elle permet d’identifier dans une protéine les positions sous sélection positive [37, 39]. Ainsi, dans le cas du gene DAZ (deleted in azoospermia), un gène probablement impliqué dans des cas de stérilité chez l’homme, l’observation que Kns était égal à Ks sur l’ensemble du gène a permis de conclure à l’absence de contrainte fonctionnelle au niveau de la séquence primaire de cette protéine. Cependant, l’estimation de Kns et de Ks à chaque codon indique qu’il y aurait en réalité des sites subissant une forte sélection négative et d’autres soumis à une sélection positive [39]. Une information complémentaire peut être apportée par l’analyse du polymorphisme à l’intérieur des espèces. La comparaison de ce polymorphisme avec les substitutions fixées entre espèces permet aussi de développer des tests de détection de la sélection [40, 41]. La recherche intensive de polymorphisme au niveau d’un nucléotide unique (SNP) dans le génome humain et quelques autres espèces, à des fins d’analyses d’association avec des maladies génétiques, donne accès à une grande quantité d’information qui permettra de mieux décrire les contraintes sélectives impliquées dans l’évolution de ce polymorphisme [42]. À ce jour, peu d’études ont été menées dans ce sens, mais plusieurs observations montrent que quelques hypothèses sous-jacentes aux modèles utilisés peuvent ne pas être toujours vérifiées. Une première hypothèse forte est que les mutations synonymes sont neutres. Elle est probablement vraie pour la plupart des gènes de mammifères, dont l’homme [43], mais la fixation des mutations synonymes pourrait parfois avoir lieu sous une importante pression de sélection [44, 45]. Par ailleurs, la probabilité d’apparition et la nature d’une mutation à une position donnée dépend de la nature des nucléotides environnants et de sa localisation sur le chromosome qui peuvent varier d’une espèce à l’autre [46]. La prise en compte de ces observations permettra de développer des méthodes plus fiables de détection de la sélection au niveau des acides aminés.
La reconstruction phylogénétique et la compréhension de l’évolution moléculaire sont deux domaines interféconds. Actuellement, les données issues du séquençage de génomes fournissent un matériel brut qui se prête très bien à une analyse bio-informatique et qui a fait grandement progresser ces deux domaines. Néanmoins, ces analyses in silico permettent essentiellement de proposer de nouvelles hypothèses, qu’il est toujours nécessaire de tester expérimentalement. Un retour à l’expérience in vitro et in vivo doit être systématique.
Glossaire
Ordre d’agglomération : Une méthode pour construire un arbre phylogénétique consiste à agglomérer les espèces progressivement. On commence par sélectionner trois espèces, pour lesquelles il n’y a qu’un arbre possible. Puis on prend une quatrième espèce et on essaie toutes les positions possibles de cette espèce dans l’arbre (ici trois) et on choisit la meilleure phylogénie contenant ces quatre espèces. Puis on recommence avec une nouvelle espèce et on continue jusqu’à ce que toutes les espèces soient ajoutées. Il y a plusieurs possibilités pour choisir l’ordre d’agglomération, par exemple, celui donné par l’utilisateur, celui fondé sur la similitude entre séquences. En réalité, le mieux est de choisir un ordre aléatoire et de répéter la procédure plusieurs fois pour contrôler l’influence de l’ordre d’agglomération sur la topologie de l’arbre final.
Maximum de parcimonie : On choisit l’arbre qui minimise le nombre de changements de nucléotides ou d’acides aminés au cours de l’évolution.
Méthodes de distances : La première étape consiste à calculer la distance évolutive (c’est-à-dire le nombre de substitutions) entre deux séquences, en général à l’aide de méthodes statistiques utilisant un modèle d’évolution des séquences. Cela aboutit à l’édification d’une matrice de distances entre toutes les paires de séquences. Différents algorithmes, comme le célèbre neighbor joining, permettent de construire rapidement un arbre à partir de cette matrice.
Transition versus transversion : On dit qu’une mutation nucléotidique est une transition lorsqu’elle transforme une purine (A ou G) en purine ou une pyrimidine (C ou T) en pyrimidine. Dans le cas contraire (purine vers pyrimidine ou pyrimidine vers purine), c’est une transversion.
Distribution gamma : C’est une fonction qui permet de modéliser la variation du taux de substitutions entre sites. Suivant la valeur du paramètre de forme, la distribution peut prendre différentes formes, en « L » (très grande hétérogénéité de vitesse entre positions, comme par exemple pour l’ARN ribosomique) ou en « cloche » (homogénéité de vitesse entre les sites, comme par exemple pour un pseudo-gène).
Attraction des longues branches : C’est un artéfact de reconstruction qui se traduit par le mauvais positionnement des séquences évoluant plus vite ou moins vite que la moyenne. En particulier, les séquences qui évoluent plus vite apparaissent d’émergence beaucoup trop précoce dans les phylogénies. C’est un artéfact très fréquemment rencontré, en particulier pour les phylogénies anciennes ou pour les phylogénies de familles multigéniques.
Références
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17 : 540–52. [Google Scholar]
- Hasegawa M, Fujiwara M. Relative efficiencies of the maximum likelihood, maximum parsimony, and neighbor-joining methods for estimating protein phylogeny. Mol Phylogenet Evol 1993; 2 : 1–5. [Google Scholar]
- Sogin ML. Early evolution and the origin of eukaryotes. Curr Opin Genet Dev 1991; 1 : 457–63. [Google Scholar]
- Van de Peer Y, Ben Ali A, Meyer A. Microsporidia: accumulating molecular evidence that a group of amitochondriate and suspectedly primitive eukaryotes are just curious fungi. Gene 2000; 246 : 1–8. [Google Scholar]
- Moreira D, Le Guyader H, Philippe H. The origin of red algae: implications for the evolution of chloroplasts. Nature 2000; 405 : 69–72. [Google Scholar]
- Chaw SM, Parkinson CL, Cheng Y, Vincent TM, Palmer JD. Seed plant phylogeny inferred from all three plant genomes: monophyly of extant gymnosperms and origin of Gnetales from conifers. Proc Natl Acad Sci USA 2000; 97 : 4086–91. [Google Scholar]
- Graur D, Hide WA, Li WH. Is the guinea-pig a rodent? Nature 1991; 351 : 649–52. [Google Scholar]
- D’Erchia A, Gissi C, Pesole G, Saccone C, Arnason U. The guinea-pig is not a rodent. Nature 1996; 381 : 597–600. [Google Scholar]
- Madsen O, Scally M, Douady CJ, et al. Parallel adaptive radiations in two major clades of placental mammals. Nature 2001; 409 : 610–4. [Google Scholar]
- Murphy WJ, Eizirik E, Johnson WE, et al. Molecular phylogenetics and the origins of placental mammals. Nature 2001; 409 : 614–8. [Google Scholar]
- Philippe H. Rodent monophyly: pitfalls of molecular phylogenies. J Mol Evol 1997; 45 : 712–5. [Google Scholar]
- Sankoff D. Gene and genome duplication. Curr Opin Genet Dev 2001; 11 : 681–4. [Google Scholar]
- Ohno S. Evolution by gene duplication. Berlin: Springer Verlag, 1970. [Google Scholar]
- Initiative TAG. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000; 408 : 796–815. [Google Scholar]
- Gu X, Wang Y, Gu J. Age distribution of human gene families shows significant roles of both large- and small-scale duplications in vertebrate evolution. Nat Genet 2002; 31 : 205–9. [Google Scholar]
- Friedman R, Hughes AL. Pattern and timing of gene duplication in animal genomes. Genome Res 2001; 11 : 1842–7. [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature 2000; 405 : 299–304. [Google Scholar]
- Perna NT, Plunkett G, Burland V, et al. Genome sequence of enterohaemorrhagic Escherichia coli O157: H7. Nature 2001; 409 : 529–33. [Google Scholar]
- Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci USA 1998; 95 : 9413–7. [Google Scholar]
- Aravind L, Tatusov RL, Wolf YI, Walker DR, Koonin EV. Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends Genet 1998; 14 : 442–4. [Google Scholar]
- Koonin EV, Makarova KS, Aravind L. Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol 2001; 55 : 709–42. [Google Scholar]
- Ragan MA. On surrogate methods for detecting lateral gene transfer. FEMS Microbiol Lett 2001; 201 : 187–91. [Google Scholar]
- Lander ES, Liton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature 2001; 409 : 860–921. [Google Scholar]
- Stanhope MJ, Lupas A, Italia MJ, et al. Phylogenetic analyses do not support horizontal gene transfers from bacteria to vertebrates. Nature 2001; 411 : 940–4. [Google Scholar]
- Doolittle WF. Phylogenetic classification and the universal tree. Science 1999; 284 : 2124–9. [Google Scholar]
- Jain R, Rivera MC, Lake JA. Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci USA 1999; 96 : 3801–6. [Google Scholar]
- Brochier C, Philippe H, Moreira D. The evolutionary history of ribosomal protein RpS14: horizontal gene transfer at the heart of the ribosome. Trends Genet 2000; 16 : 529–33. [Google Scholar]
- Brochier C, Bapteste E, Moreira D, Philippe H. Eubacterial phylogeny based on translational apparatus proteins. Trends Genet 2000; 18 : 1–5. [Google Scholar]
- Matte-Tailliez O, Brochier C, Forterre P, Philippe H. Archaeal phylogeny based on ribosomal proteins. Mol Biol Evol 2002; 19 : 631–9. [Google Scholar]
- Lopez P, Casane D, Philippe H. Heterotachy, an important process of protein evolution. Mol Biol Evol 2002; 19 : 1–7. [Google Scholar]
- Lockhart PJ, Larkum AW, Steel M, Waddell PJ, Penny D. Evolution of chlorophyll and bacteriochlorophyll: the problem of invariant sites in sequence analysis. Proc Natl Acad Sci USA 1996; 93 : 1930–4. [Google Scholar]
- Dayhoff MO. Atlas of protein sequence and structure (supplement 3, 1978). Washington: National Biomedical Research Foundation, 1979. [Google Scholar]
- Goldman N, Thorne JL, Jones DT. Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 1998; 149 : 445–58. [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge: Cambridge University Press, 1983. [Google Scholar]
- Li WH. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol 1993; 36 : 96–9. [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen AM. Codonsubstitution models for heterogeneous selection pressure at amino acid sites. Genetics 2000 ; 155 : 431–49. [Google Scholar]
- Suzuki Y, Gojobori T. A method for detecting positive selection at single amino acid sites. Mol Biol Evol 1999 ; 16 : 1315–28. [Google Scholar]
- Wyckoff GJ, Wang W, Wu CI. Rapid evolution of male reproductive genes in the descent of man. Nature 2000 ; 403 : 304–9. [Google Scholar]
- Bielawski JP, Yang Z. Positive and negative selection in the daz gene family. Mol Biol Evol 2001 ; 18 : 523–9. [Google Scholar]
- Hudson RR, Kreitman M, Aguade M. A test of neutral molecular evolution based on nucleotide data. Genetics 1987 ; 116 : 153–9. [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature 1991 ; 351 : 652–4. [Google Scholar]
- Gaudieri S, Dawkins RL, Habara K, Kulski JK, Gojobori T. SNP profile within the human major histocompatibility complex reveals an extreme and interrupted level of nucleotide diversity. Genome 2000 ; 10 : 1579–86. [Google Scholar]
- Sharp PM, Cowe E, Higgins DG, et al. Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens: a review of the considerable within-species diversity. Nucleic Acids Res 1988 ; 16 : 8207–11. [Google Scholar]
- Eyre-Walker A. Evidence of selection on silent site base composition in mammals: potential implications for the evolution of isochores and junk DNA. Genetics 1999 ; 152 : 675–83. [Google Scholar]
- Dufour C, Casane D, Denton D, et al. Human-chimpanzee DNA sequence variation in the four major genes of the renin angiotensin system. Genomics 2000 ; 69 : 14–26. [Google Scholar]
- Casane D, Boissinot S, Chang BH, Shimmin LC, Li W. Mutation pattern variation among regions of the primate genome. J Mol Evol 1997 ; 45 : 216–26. [Google Scholar]
Liste des figures
|
Figure 1. Information et bruit lors de la construction des phylogénies moléculaires. Sur cet arbre phylogénétique est représentée l’évolution de trois positions, i, j et k d’une protéine. La position i n’a subi qu’une seule substitution, se traduisant par le changement de l’acide aminé lysine (K) en tyrosine (Y). Si cette mutation s’est fixée dans l’ancêtre commun des espèces 9-15, on peut envisager le regroupement des espèces 9-15 à l’exclusion de toutes les autres, car ces espèces sont les seules à posséder un Y à la position i. Dans ce cas, le signal apporté par la position i est peu bruité. Malheureusement, une mutation peut aussi aisément créer du « bruit », ce qui peut amener à des artéfacts de reconstruction: c’est le cas des positions j et k où deux substitutions ont eu lieu, une réversion en j, et une convergence en k. Imaginons par exemple que l’ancêtre commun des espèces 22-27 fixe une mutation qui change la valine (V) en isoleucine (I) à la position j, mais que l’ancêtre de l’espèce 27 fixe une mutation qui change I en V (ce que l’on appelle une réversion), alors l’espèce 27 va être illégitimement exclue du groupe 22-27. De même, si les ancêtres communs des groupes 4-6 et 16-21 fixent, de façon indépendante et à cette même position k, une mutation qui change l’acide aspartique (D) en acide glutamique (E) (ce que l’on appelle une convergence), alors les espèces 4-6 seraient indûment groupées avec les espèces 16-21. |
| Dans le texte | |
|
Figure 2. Phylogénie du gène D1 codant pour un récepteur de la dopamine. Cet arbre a été « raciné » sur la myxine (un poisson dépourvu de mâchoires), c’est-à-dire que l’on a décidé, sur la base d’informations extérieures (ici paléontologiques et morphologiques) de placer le début de l’histoire de ces séquences entre la myxine et les gnathostomes (poissons à mâchoires). En effet, à partir des seules données moléculaires, il est très difficile de positionner le début de l’histoire (à moins de faire de nombreuses hypothèses). Le gène D1 s’est dupliqué en quatre copies chez l’ancêtre des vertébrés (notées ici A, B, C et D), qu’on appelle des paralogues (voir glossaire). De plus, le paralogue A s’est luimême dupliqué au sein des poissons, ce qui prouve que de tels événements sont fréquents. La résolution de l’arbre n’est pas bonne (les branches significativement robustes sont représentées en rouge), sans doute à cause du petit nombre de positions (~ 300). On notera que la phylogénie n’est pas parfaitement conforme à celle des espèces, comme le montre la non-monophylie des poissons pour D1A et D1B. Un scénario possible pour l’évolution de ce récepteur est que les quatre copies ont été engendrées par les deux événements hypothétiques de tétraploïdisation, que certaines ont été perdues dans plusieurs lignées de vertébrés et que d’autres événements de duplication (en tandem ou par tétraploïdisation) ont eu lieu chez les poissons téléostéens. En particulier, on notera l’absence des gènes D1C et D1D chez les mammifères, qui n’est pas due à un problème d’échantillonnage mais à deux pertes. Pour ce gène impliqué dans le système nerveux central, contrairement aux idées préconçues, les poissons ont plus de gènes que les mammifères. La barre correspond à un nombre moyen de 0,1 changement par position. |
| Dans le texte | |
|
Figure 3. Estimation de la sélection au niveau moléculaire. Dans cet exemple simplifié, on considère quatre espèces actuelles (A, B, C, D) et quatre codons. Après avoir inféré les relations phylogénétiques entre ces espèces, on infère les séquences chez les trois ancêtres communs. Ensuite, pour chaque codon, on compte le nombre de substitutions synonymes (s), c’est-à-dire qui ne change pas l’acide aminé (indiqué en vert) et non-synonymes (ns), c’est-à-dire qui change l’acide aminé (indiqué en rouge). Dans le cas du premier codon, il y a eu trois mutations synonymes (s = 3) et une mutation non-synonyme (ns = 1). Comme on s’intéresse à la fréquence des changements, il faut corriger par le nombre potentiel de mutations synonymes (Ns) et non-synonymes (Nns). Par exemple, pour la valine (codé par GTN), il y a trois mutations synonymes possibles et six non-synonymes, alors que, pour le tryptophane (codé uniquement par UGG), il n’y a aucune mutation synonyme. Grâce à des méthodes plus ou moins sophistiquées, on calcule Ks (s/Ns) et Kns (ns / Nns). Ks est un estimateur du taux de mutation, Kns est un estimateur du taux de changement des acides aminés. Pour le premier codon, Kns est inférieur à Ks, ce qui indique une sélection négative qui empêche la fixation de certains changements d’acides aminés. Pour le deuxième codon, Kns est supérieur à Ks, ce qui indique une sélection positive qui favorise des changements d’acides aminés. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.