")
")
| Issue |
Med Sci (Paris)
Volume 17, Number 12, Décembre 2001
|
|
|---|---|---|
| Page(s) | 1289 - 1296 | |
| Section | Articles de Synthèse | |
| DOI | https://doi.org/10.1051/medsci/200117121289 | |
| Published online | 15 décembre 2001 | |
Étiologie moléculaire des rachitismes vitamino-dépendants héréditaires
Hereditary defects in vitamin D-deficiency rickets
Unité de génétique, Hôpital Shriners pour enfants, 1529, avenue Cedar, Mont-réal, Québec H3G 1A6, Canada
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La vitamine D subit deux transformations métaboliques conduisant à la synthèse de sa forme hormonale, la 1,25-dihydroxyvitamine D. Des mutations dans le gène du cytochrome P450, 25-hydroxyvitamine D-1-α-hydroxylase (1α-OHase), sont responsables d’une forme de rachitisme vitamino-dépendant héréditaire, nommée PDDR (pseudovitamin D deficiency rickets) . Vingt-six mutations différentes, incluant des duplications et des délétions modifiant le cadre de lecture, et des mutations ponctuelles affectant la séquence de la protéine, l’épissage, ou conduisant à l’arrêt prématuré de la traduction, ont été détectées chez les patients de PDDR. Nous avons créé un modèle animal de PDDR en inactivant le gène 1 α -OHase chez des souris. Cet article présente le phénotype des souris mutantes, résume l’état actuel des connaissances concernant l’expression et la fonction de la 1α OHase , et discute des nouvelles perspectives de recherche sur le métabolisme de la vitamine D.
Abstract
Vitamin D is a key regulator of mineral homeostasis and bone development. Two types of hereditary defects in vitamin D metabolism cause rickets. The first one involves mutations in the vitamin D receptor, leading to hereditary vitamin D-resistant rickets. The second type of defects involves mutations in the cytochrome P450 enzyme, 25-hydroxyvitamin D1-alpha-hydroxylase (1α-OHase), and causes pseudo-vitamin D deficiency rickets (PDDR). Twenty-six distinct mutations, including duplications, deletions, missense and nonsense mutations have been identified in patients with PDDR. We have engineered an animal model of PDDR through targeted inactivation of the 1α-OHase gene in mice. This review presents the phenotype of the 1α-OHase-ablated mice, and provides an overview of the future avenues of research concerning the control of the expression and the physiological role of the 1α-OHase enzyme.
© 2001 médecine/sciences - Inserm / SRMS
La vitamine D est essentielle au contrôle de l’homéostasie des minéraux et, par conséquent, au développement osseux. Toute perturbation du système endocrinien de la vitamine D peut provoquer un rachitisme ou une ostéomalacie. Deux formes de rachitisme vitaminodépendant sont héréditaires. La première, nommée HVDRR (hereditary vitamin D resistant rickets), est due à des mutations dans le gène codant pour le récepteur de la vitamine D (revue récente dans [1]). L’autre, nommée PDDR (pseudovitamin D deficiency rickets), est le résultat de mutations dans le gène codant pour l’enzyme responsable de la synthèse de la forme active de la vitamine D, la 25-hydroxyvitamine D-1a-hydroxylase (1α-OHase). Le clonage récent du gène de la 1α-OHase a grandement contribué à la compréhension de l’étiologie moléculaire des rachitismes PDDR. Nous présenterons un bref aperçu du métabolisme de la vitamine D avant de discuter des nouvelles voies de recherche ouvertes par le clonage du gène de la 1α-OHase.
Métabolisme de la vitamine D
La majeure partie des besoins en vitamine D peut être comblée de façon endogène par exposition au soleil. Un apport alimentaire provenant d’un régime incluant du poisson, des légumes et des céréales complète lesbesoins en vitamine. Les animaux et les poissons synthétisent la vitamine D3 ;les plantes et les levures synthétisent la vitamine D2. Les quelques différences de structure chimique entre les deux molécules n’affectent pas leur fonction et le terme générique de vitamine D sera ensuite adopté.
La synthèse de la vitamine D est amorcée à la suite de l’exposition de la peau aux rayons du soleil. Les rayons UVB pénètrent les kératinocytes et lysent le 7-déshydrocholestérol en pro-vitamine D, une molécule instable qui est rapidement transformée en vitamine D. La vitamine D se lie à une globuline de transport, la DBP (vitamin D binding protein), pour arriver au foie où l’enzyme vitamine D-25-hydroxylase (CYP27), un cytochrome P450, la convertit en 25-hydroxyvitamine D [25(OH)D], un métabolite inactif. Toujours liée à la DBP, la 25(OH)D est transportée vers le rein dans lequel la 1α-OHase attache un groupement hydroxyle en position 1a pour engendrer la forme active de la vitamine, la 1α,25-dihydroxyvitamine D [1,25(OH)2D] [2]. Cette dernière se lie à son récepteur (VDR, vitamin D receptor), un facteur de transcription de la famille des récepteurs nucléaires qui contrôle l’expression des gènes cibles de la vitamine D (figure 1).
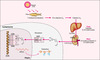 |
Figure 1. Biosynthèse de la 1,25(OH)2D3. La vitamine D (ici D3 puisque le schéma s’applique à l’homme), synthétisée suite à l’irradiation de la peau par les rayons solaires, est convertie en 25-hydroxyvitamine D3 [25(OH)D3] par la vitamine D 25-hydroxylase au niveau du foie. Ce métabolite inactif est ensuite métabolisé en 1α,25-dihydroxyvitamine D3 [1,25(OH)2D3] à la suite de l’action de l’enzyme rénale, la 25-hydroxyvitamine D-1α-hydroxylase (1α-OHase). L’hormone 1,25(OH)2D3 se lie ensuite à son récepteur nucléaire, le VDR ( vitamin D receptor), un récepteur nucléaire de type II qui forme un dimère actif avec son partenaire, le RXR (retinoid X receptor) , et module la transcription de gènes cibles. Le ligand du RXR, l’acide 9-cisrétinoïque, n’est pas illustré. La cause moléculaire des rachitismes PDDR (pseudo vitamin D deficiency rickets) , – des mutations dans le gène codant pour la 1 α-OHase – est illustrée par la flèche interrompue au niveau du rein. L’autre forme de rachitisme héréditaire vitamino-dépendant, HVD-RR (hereditary vitamin D resistant rickets) , est due à des mutations affectant la structure ou la fonction du VDR, soit au niveau du domaine de liaison du ligand (LBD, ligand binding domain ), soit au niveau du domaine de liaison à l’ADN (DBD, DNA binding domain) . |
Nommée vitamine pour des raisons historiques liées au fait que des préparations d’huile de foie de morue pouvaient guérir du rachitisme, l’étude du métabolisme de la vitamine D et de la modulation hormonale de la synthèse de sa forme active, la 1,25(OH)2D, révèle que ce métabolite est en fait une hormone. Le rôle physiologique majeur de la 1,25(OH)2D est de stimuler l’absorption du calcium et du phosphate dans le duodénum et de stimuler la réabsorption du calcium et du phosphate par le néphron [2]. De cette façon, la 1,25(OH)2D joue un rôle majeur dans l’homéostasie des minéraux et assure un apport suffisant d’ions pour une minéralisation correcte de la matrice osseuse.
Plusieurs études ont suggéré que la 1,25(OH)2D aurait une action directe sur différents types cellulaires tel l’ostéoblaste, les macrophages ou les kératinocytes [2]. Le phénotype dessouris ayant une mutation ciblée dans le gène du VDR démontre que le rôle principal de la 1,25(OH)2D concerne l’homéostasie des minéraux [3, 4]. En effet, le développement embryonnaire de ces souris est normal, et elles survivent jusqu’à l’âge de 6 mois sans traitement. A la suite du sevrage, elles développent une hypocalcémie et une hyperparathyroïdie secondaire [3, 4]. Cependant, des études plus approfondies à l’aide de ces puissants modèles génétiques pourraient permettre de confirmer sans équivoque certaines des actions de la 1,25(OH)2D sur des tissus cibles autres que le rein et l’intestin. Déjà, un rôle essentiel du VDR et, par inférence, de la 1,25(OH)2D, a été mis en évidence dans le cycle folliculaire pileux [5].
Rachitisme versus ostéomalacie
Le terme « rachitisme » est souvent employé pour définir toute anomalie de minéralisation du squelette. En réalité, le rachitisme affecte seulement la plaque de croissance (voir l’article de P. Ducy, p. 1242 de ce numéro), et l’accumulation de matrice non minéralisée sur d’autres sites est correctement appelée ostéomalacie. Un individu en période de croissance peut donc souffrir à la fois de rachitisme et d’ostéomalacie, alors qu’un adulte ne montrera que des lésions ostéomalaciques.
Toute perturbation du métabolisme de la vitamine D peut provoquer un rachitisme et/ou une ostéomalacie:exposition insuffisante aux rayons solaires ; carence alimentaire ; maladies du foie ou des reins; déficit en vitamine D-25-hydroxylase hépatique déficit en 1α-OHase rénale ; inactivité du VDR. Parmi toutes ces causes, seules le déficit en 1α-OHase rénale (PDDR) et l’inactivité du VDR (HVDRR) ont été associées à des mutations dans des gènes spécifiques.
Physiopathologie des rachitismes PDDR
Les symptômes des patients atteints de rachitisme PDDR ressemblent aux symptômes classiques d’individus carencés en vitamine D: croissance ralentie, hypotonie, troubles moteurs et évidence radiologique de rachitisme (hypominéralisation et apparence diffuse et élargie de la zone de calcification sous la plaque de croissance). Les nouveau-nés atteints ont une apparence normale car les symptômes se manifestent à partir du troisième trimestre. Au niveau sérologique, les niveaux de calcium sont anormalement bas, ce qui provoque une hyperparathyroïdie secondaire. Le calcium urinaire est bas alors que la concentration fécale est élevée, conséquence de la malabsorption intestinale. La phosphatémie peut être normale ou basse.
La caractéristique principale des mesures sérologiques chez les patients atteints de rachitisme PDDR concerne les niveaux circulants des métabolites de la vitamine D. Alors que les niveaux sériques de 25(OH)D sont normaux, les niveaux circulants de 1,25(OH)2D sont non détectables ou à la limite inférieure des dosages.
Cette caractéristique a rapidement orienté les recherches vers une cause moléculaire des rachitismes PDDR impliquant l’enzyme responsable de la synthèse de la 1,25(OH)2D: la 1α-OHase. Des mesures de l’activité enzymatique dans des cellules déciduales, qui expriment l’enzyme, ont confirmé l’absence d’activité 1α-OHase chez les [6]. Les conclusions de ces études biochimiques classiques ont été confirmées par le clonage du gène codant pour la 1α-OHase [7–10]. Non seulement le gène isolé a-t-il été cartographié au locus de la maladie, mais des mutations inactivatrices ont été identifiées chez les patients et les individus hétérozygotes porteurs de la maladie (voir plus loin).
Traitement
Le traitement approprié des rachitismes de type PDDR est l’apport de 1,25(OH)2D3 [11]. Très rapidement après le début du traitement, les paramètres sériques et radiologiques sont normalisés. De même, la myopathie disparaît rapidement. Il est important de fournir parallèlement un apport calcique suffisant au début du traitement afin de permettre une bonne guérison des lésions osseuses. L’apport calcique doit être contrôlé par la mesure de l’excrétion urinaire. Une excrétion anormalement élevée peut provoquer des néphrocalcinoses et une surveillance adéquate de la fonction et de la structure rénale est recommandé.
A l’époque où la 1,25(OH)2D3 n’était pas disponible commercialement, plusieurs cliniciens ont démontré l’efficacité d’un traitement avec l’analogue 1α(OH)D3 [12]. Cette molécule peut être métabolisée en 1,25(OH)2D3 puisque la 25-hydroxylase hépatique fonctionne normalement chez les patients PDDR. Cette thérapeutique est encore en usage dans plusieurs pays. Cependant, les coûts de production de l’analogue ne procurent aucun avantage financier à cette forme de traitement.
Clonage de la 1α-OHase
Il a été difficile de cloner le gène codant pour la 1α-OHase en raison de son faible niveau d’expression. D’ingénieuses techniques ont dû être mises au point afin d’isoler les premiers ADNc. La séquence a été obtenue chez le rat par criblage à stringence réduite d’une banque d’ADNc de reins d’animaux carencés en vitamine D, à l’aide d’une sonde provenant d’un gène homologue, la 25-hydroxyvitamine D-24-hydroxylase (24-OHase) [8]. Une stratégie similaire, fondée sur des amorces dégénérées dérivées de la séquence de la 24-OHase et l’utilisation de la PCR (polymerase chain reaction), ont conduit au clonage de l’ADNc humain [10] et confirmé la séquence du gène chez le rat [7]. Quant à l’ADNc murin, il a été obtenu en utilisant un élégant modèle génétique et une technologie originale. Les souris portant une mutation inactivatrice du VDR ont des niveaux d’expression élevés de la 1α-OHase dus à l’absence de rétrocontrôle négatif; elles ont donc été une source privilégiée pour la construction d’une banque d’ADNc enrichie pour la séquence de la 1α-OHase. Le clonage a utilisé une technique d’expression de cellules de mammifères dépendante de l’expression conditionnelle d’un gène rapporteur à la suite de la synthèse de 1,25(OH)2D3 par la cellule transfectée [9].
Les clones d’ADNc ont rapidement permis d’isoler des clones génomiques humains. A l’aide de telles sondes, le gène humain a été cartographié sur le locus de la maladie, préalablement identifié par analyse de liaison [13, 14].
Mutations chez les patients atteints de PDDR
Vingt-six mutations différentes ont été détectées chez les 38 patients atteints de PDDR analysés jusqu’à présent (Tableau I) [10, 15–19]. Plusieurs types de mutations ont été identifiés : duplications et délétions modifiant le cadre de lecture, et mutations ponctuelles affectant la séquence de la protéine (missense mutations), l’épissage, ou conduisant à l’arrêt prématuré de la traduction (nonsense mutation). Des mutations ont été détectées dans chacun des 9 exons du gène, de même que dans l’intron 3, mutation qui modifie l’épissage de l’ARNm (figure 2). Une mutation spécifique a été détectée avec une plus grande fréquence : il s’agit d’une duplication de 7 paires de bases qui change le cadre de lecture au codon 443 et provoque l’arrêt prématuré de la traduction au codon 465 (Tableau I). Cette mutation est présente chez 11 patients issus de 10 familles non apparentées [15–17]. Une caractéristique de l’étiologie moléculaire de la maladie semble être la présence de mutations hétérozygotes : chez 15 des 36 cas publiés, une mutation différente a été identifiée sur chacun des deux allèles [10, 15–19].
 |
Figure 2. Mutations chez les patients atteints de rachitisme PDDR (pseudo-vitamin D deficiency rickets). La structure du gène de la 25-hydroxyvitamine D-1a-hydroxylase (1a-OHase) est représentée de façon schématique. Les exons sont numérotés de 1 à 9; les régions rouges correspondent aux séquences non-traduites en 5’ et en 3’, respectivement. Les 26 mutations identifiées jusquàa présent chez des patients atteints de rachitisme PDDR sont représentées au-dessus et en dessous de la carte du gène. Les nombres indiquent le nucléotide ou l’acide aminé muté, selon le cas. Δ : délétion; IVS3 + 1g->a: mutation dans l’intron 3 (intervening sequence 3) affectant l’epis-sage; 7 bp dupl: duplication de 7 paires de bases; 2 pb dupl: duplication de 2 paires de bases. |
Mutations du gène de la 25-hydroxyvitamine D-1α-hydroxylase chez les patients atteints de rachitisme.
Il est facile de comprendre l’impact des mutations affectant le cadre de lecture et produisant une protéine tronquée inactive. L’impact des mutations ponctuelles pourrait s’avérer plus subtil. Une première analyse fondée sur la synthèse de 1,25(OH)2D par des cellules transfectées, à l’aide de vecteurs d’expression, avec des séquences mutées a révélé une perte complète d’activité pour chacun des mutants testés [10, 15–19]. Wang et al. [16] ont tenté d’expliquer la perte de fonction en s’appuyant sur la structure cristallographique d’un cytochrome P450 apparenté. Chacune des mutations identifiées semble affecter un domaine essentiel de la protéine tel le site d’entrée ou de liaison du substrat, ou le site de liaison du groupement prosthétique (heme binding domain). Néanmoins, il demeurait possible que certaines mutations ponctuelles puissent provoquer des pertes d’activité enzymatique modérées qui ne pourraient être détectées à l’aide des méthodes utilisées jusqu’à présent. Cette éventualité revêt une importance particulière quand elle est liée au phénotype de patients atteints de forme modérée de rachitisme PDDR. Chez ces individus, les niveaux circulants de 1,25(OH)2D sont faibles mais détectables [20]. Le groupe de Shigeaki Kato est parvenu à purifier la forme active de la 1α-OHase recombinante exprimée chez E. coli et à étudier la cinétique enzymatique de la protéine recombinante [21, 22]. En appliquant la même technique aux différents mutants ponctuels de la 1α-OHase, et en particulier à une mutation détectée chez un patient atteint d’une forme modérée de PDDR, cette équipe a démontré de façon convaincante que les mutations ponctuelles provoquent une perte totale d’activité enzymatique [18]. Bien que la persistance d’une activité enzymatique résiduelle des protéines mutées ou la présence d’un autre gène impliqué dans l’hydroxylation de la vitamine D en position 1α ne puissent être formellement exclues, l’explication la plus logique du phénotype PDDR modéré semble impliquer un apport alimentaire de 1,25(OH)2D (par exemple dans le lait de vache et le poisson) [23, 24], ou encore la détection croisée de métabolites inactifs de la vitamine D par la méthode de dosage de la 1,25(OH)2D.
Recherches futures
Contrôle de l’expression
L’expression du gène de la 1α-OHase est soumise à une modulation complexe impliquant les niveaux circulants de calcium et de phosphate, la calcitonine, la parathormone (PTH), et son propre produit enzymatique, la 1,25(OH)2D [2, 25–28]. Les mécanismes moléculaires responsables de ces réponses commencent à être connus. La réponse à la PTH requiert l’activité de la protéine kinase A (PKA) et la formation d’AMPc [25, 27]. Des éléments de réponse à l’AMPc (cyclic AMP response elements, CRE) ont été identifiés dans le promoteur proximal du gène codant pour la 1α-OHase [25]. Les prochaines études s’appliqueront à démontrer la fonctionnalité des éléments identifiés.
Un aspect fascinant du contrôle de l’expression du gène de la 1α-OHaseconcerne sa régulation négative par la 1,25(OH)2D. En effet, l’analyse d’un fragment du promoteur du gène de la 1α-OHase répondant à la 1,25(OH)2D ne révèle aucun élément de réponse à la vitamine D (vitamin D response element, VDRE, ou negative vitamin D response element, nVDRE) [25]. L’étude approfondie du fragment du promoteur qui contrôle la réponse à la 1,25(OH) 2D a permis d’isoler un nouveau membre de la famille des régulateurs transcriptionels bHLH (basic domain-helix-loophelix). Ce facteur bHLH serait impliqué dans l’expression basale du gène 1 αOHase, et une interaction protéine-protéine avec le VDR serait responsable de l’inhibition transcriptionnelle par la 1,25(OH) 2D [29]. Ces découvertes de l’équipe de S. Kato suggèrent des mécanismes de contrôle jusqu’à présent insoupçonnés et identifient une nouvelle cible potentielle d’intervention thérapeutique.
Modèle animal : inactivation ciblée
Afin de mettre au point un modèle animal de rachitisme PDDR, nous avons inactivé le gène 1 αOHase par recombinaison homologue dans des cellules embryonnaires (ES). La stratégie utilisée fait appel au système de recombinase Cre-loxP du bactériophage P1 [30]. Ainsi, à partir du même événement de recombinaison, nous avons pu mettre au point des mutants complets (inactivation danstous les tissus à la suite de l’activité de la recombinase Cre dans les cellules ES) et des mutants conditionnels (inactivation spécifique après croisement avec des souris transgéniques exprimant Cre dans un tissu donné). Le phénotype des mutants complets est une copie du phénotype PDDR chez l’homme [31]. La croissance des animaux mutants est ralentie et les radiographies osseuses confirment le rachitisme (figure 3). De même, tous les paramètres sériques et histologiques reproduisent la pathologie humaine. Si les animaux mutants ne sont pas traités par thérapie de remplacement avec la 1,25(OH)2D, ils meurent au début de l’âge adulte (figure 4). Ce modèle animal pourra être utilisé afin de tester d’autres approches de sauvetage du phénotype par altération du régime. Les mutants pourraient aussiâtre utiles afin de mesurer l’activité calcémique d’analogues de la 1,25(OH)2D en l’absence de l’hormone endogène.
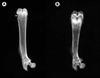 |
Figure 3. Radiographies du fémur d’animaux déficients pour le gène de la 25-hydroxyvitamine D-1α-hydroxylase (1α-OHase). A. Fémur d’un animal hétérozygote. B. Fémur d’un homozygote mutant pour la 1α-OHase. Notez l’élargissement de la plaque de croissance et la minceur du cortex du fémur mutant, démontrant la présence de rachitisme et d’ostéomalacie. |
 |
Figure 4. Survie des homozygotes mutants pour le gène de la 25-hydroxyvitamine D-1α-hydroxylase (1α-OHase). Les souris déficientes pour le gène 1α-OHase ont é t é sevrées à l’âge de trois semaines et alimentées par un régime normal. Dans ces conditions, les animaux meurent entre la 11e et la 16e semaine de vie. |
Rôle autocrine/paracrine
Bien que le site majeur d’expression de la 1α-OHase soit le rein, il a été démontré que le gène est exprimé dans d’autres types cellulaires, tels que les ostéoblastes, les chondrocytes, les kératinocytes, les macrophages, et certains types de neurones [2]. Combinées aux résultats démontrant un rôle possible de la 1,25(OH)2D sur la différenciation ou la fonction de ces cellules [2, 32, 33], ces observations suggèrent un contrôle autocrine/paracrine possible de l’activité hormonale de la vitamine D dans ses tissus cibles. La technologie Cre-loxP, utilisée pour la création des mutants conditionnels, nous permettra d’étudier l’importance physiologique des fonctions suggérées.
En croisant les souris 1α-OHase-loxP avec des souris transgéniques exprimant Cre sous le contrôle du promoteur ostéocalcine [34], il sera possible d’inactiver la 1α-OHase spécifiquement dans les ostéoblastes différenciés. Cette approche permettra d’étudier le rôle direct de la 1,25(OH)2D sur la fonction osseuse. Des travaux récents utilisant des mutations de gain de fonction ou de perte de fonction du VDR dans l’os suggèrent un rôle de la 1,25(OH)2D distinct de son implication dans l’homéostasie des minéraux [35, 36]. La fonction paracrine de la 1,25(OH)2D suggérée dans la maturation des chondrocytes pourra aussi être testée en croisant les animaux 1a-OHase-loxP avec des souris transgéniques exprimant Cre sous le contrôle des séquences régulatrices du gène du collagène de type II.
Le gène 1αOHase pourra être inactivé dans les kératinocytes en croisant les souris 1α-OHase- loxP avec des souris transgéniques kératine-Cre [37]. Ces études pourraient confirmer le rôle important de la 1,25(OH)2D dans la différenciation de la peau, déjà mis en évidence chez les souris déficientes en VDR. En effet, un régime riche en calcium peut rétablir la calcémie et corriger tous les aspects du phénotype de ces animaux mutants, sauf la différenciation de la peau et le cycle folliculaire pileux [5]. La recombinase Cre pourra être exprimée sous le contrôle du promoteur du gène macrophage scavenger receptor A [38] afin d’inactiver la 1α-OHase dans les macrophages. Plusieurs études démontrent que la 1,25(OH)2D stimule la fonction des macrophages en augmentant leur activité chimiotactique et phagocytaire [32]. Il est possible que l’inactivation ciblée de la 1a-OHase dans les macrophages diminue la capacité des animaux mutants de lutter contre des infections microbiennes.
Perspectives
Les puissantes techniques de la génétique moléculaire vont bientôt permettre de confirmer ou d’infirmer les rôles physiologiques suggérés pour l’hormone 1,25(OH)2D. Déjà, les travaux récents ont confirmé l’implication du gène de la 1a-OHase dans l’étiologie moléculaire des rachitismes PDDR, ont démontré un rôle essentiel de la 1,25(OH)2D dans le cycle folliculaire pileux, et ont identifié de nouveaux mécanismes de modulation transcriptionnelle impliquant la 1,25(OH)2D. L’inactivation conditionnelle du gène de la 1a-OHase dans les tissus cibles de la 1,25(OH)2D permettra sans doute de démontrer des fonctions importantes de l’hormone, s’ajoutant à son rôle essentiel dans le contrôle de l’homéostasie des minéraux
Remerciements
Nous remercions Guylaine Bédard pour la préparation des illustrations. Les travaux provenant du laboratoire des auteurs ont été subventionnés par les Shriners d’Amérique du Nord. R.Saint-Arnaud est un chercheur-boursier du Fonds de la Recherche en Santé du Québec.
Références
- Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25- dihydroxyvitamin D-resistant rickets. Endocrinol Rev 1999; 20 : 156–88. [Google Scholar]
- Feldman D, Glorieux FH, Pike JW. Vitamin D. San Diego : Academic Press, 1997. [Google Scholar]
- Li YC, Pirro AE, Amling M, et al. Targeted ablation of the vitamin D receptor : an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci USA 1997; 94 : 9831–5. [Google Scholar]
- Yoshizawa T, Handa Y, Uematsu Y, et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet 1997; 16 : 391–6. [Google Scholar]
- Li YC, Amling M, Pirro AE, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology 1998; 139: 4391–6. [Google Scholar]
- Glorieux FH, Arabian A, Delvin EE. Pseudo-vitamin D deficiency : absence of 25-hydroxyvitamin D 1 alpha- hydroxylase activity in human placenta decidual cells. J Clin Endocrinol Metab 1995; 80 : 2255–8. [Google Scholar]
- Shinki T, Shimada H, Wakino S, et al. Cloning and expression of rat 25-hydroxyvitamin D3-1alpha-hydroxylase cDNA. Proc Natl Acad Sci USA 1997; 94 : 12920–5. [Google Scholar]
- St-Arnaud R, Messerlian S, Moir JM, Omdahl JL, Glorieux FH. The 25-hydroxyvitamin D 1-alpha-hydroxylase gene maps to the pseudovitamin D-deficiency rickets (PDDR) disease locus. J Bone Miner Res 1997; 12 : 1552–9. [Google Scholar]
- Takeyama K, Kitanaka S, Sato T, Kobori M, Yanagisawa J, Kato S. 25-Hydroxyvitamin D3 1alpha-hydroxylase and vitamin D synthesis. Science 1997; 277 : 1827–30. [Google Scholar]
- Fu GK, Lin D, Zhang MY, et al. Cloning of human 25-hydroxyvitamin D-1 alphahydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol 1997; 11 : 1961–70. [Google Scholar]
- Delvin EE, Glorieux FH, Marie PJ, Pettifor JM. Vitamin D dependency : replacement therapy with calcitriol ? J Pediatr 1981; 99 : 26–34. [Google Scholar]
- Reade TM, Scriver CR, Glorieux FH, et al. Response to crystalline 1alpha-hydroxyvitamin D3 in vitamin D dependency. Pediatr Res 1975; 9 : 593–9. [Google Scholar]
- Labuda M, Labuda D, Korab-Laskowska M, et al. Linkage disequilibrium analysis in young populations : pseudo-vitamin D- deficiency rickets and the founder effect in French Canadians. Am J Hum Genet 1996; 59 : 633–43. [Google Scholar]
- Labuda M, Morgan K, Glorieux FH. Mapping autosomal recessive vitamin D dependency type I to chromosome 12q14 by linkage analysis. Am J Hum Genet 1990; 47 : 28–36. [Google Scholar]
- Yoshida T, Monkawa T, Tenenhouse HS, et al. Two novel 1alpha-hydroxylase mutations in French-Canadians with vitamin D dependency rickets type I1. Kidney Int 1998; 54 : 1437–43. [Google Scholar]
- Wang JT, Lin CJ, Burridge SM, et al. Genetics of vitamin D 1alpha-hydroxylase deficiency in 17 families. Am J Hum Genet 1998; 63 : 1694–702. [Google Scholar]
- Smith SJ, Rucka AK, Berry JL, et al. Novel mutations in the 1alpha-hydroxylase (P450c1) gene in three families with pseudovitamin D-deficiency rickets resulting in loss of functional enzyme activity in bloodderived macrophages. J Bone Miner Res 1999; 14 : 730–9. [Google Scholar]
- Kitanaka S, Murayama A, Sakaki T, et al. No enzyme activity of 25-hydroxyvitamin D3 1alpha-hydroxylase gene product in pseudovitamin D deficiency rickets, including that with mild clinical manifestation. J Clin Endocrinol Metab 1999; 84 : 4111–7. [Google Scholar]
- Kitanaka S, Takeyama K, Murayama A, et al. Inactivating mutations in the 25-hydroxyvitamin D3 1alpha-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med 1998; 338 : 653–61. [Google Scholar]
- Balsan S. Hereditary pseudo-deficiency rickets or vitamin D-dependency type I. In : Glorieux FH, ed. Rickets. New York : Raven Press, 1991 : 155–63. [Google Scholar]
- Sakaki T, Sawada N, Takeyama K, Kato S, Inouye K. Enzymatic properties of mouse 25-hydroxyvitamin D3 1 alpha-hydroxylase expressed in Escherichia coli . Eur J Biochem 1999; 259 : 731–8. [Google Scholar]
- Sawada N, Sakaki T, Kitanaka S, Takeyama K, Kato S, Inouye K. Enzymatic properties of human 25-hydroxyvitamin D3 1alpha-hydroxylase coexpression with adrenodoxin and NADPH-adrenodoxin reductase in Escherichia coli. Eur J Biochem 1999; 265 : 950–6. [Google Scholar]
- Takeuchi A, Okano T, Kobayashi T. The existence of 25-hydroxyvitamin D3-1 alphahydroxylase in the liver of carp and bastard halibut. Life Sci 1991; 48 : 275–82. [Google Scholar]
- Hollis BW, Roos BA, Draper HH, Lambert PW. Vitamin D and its metabolites in human and bovine milk. J Nutr 1981; 111 : 1240–8. [Google Scholar]
- Brenza HL, Kimmel-Jehan C, Jehan F, et al. Parathyroid hormone activation of the 25-hydroxyvitamin D3-1alpha-hydroxylase gene promoter. Proc Natl Acad Sci USA 1998; 95 : 1387–91. [Google Scholar]
- Murayama A, Takeyama K, Kitanaka S, Kodera Y, Hosoya T, Kato S. The promoter of the human 25-hydroxyvitamin D3 1 alpha-hydroxylase gene confers positive and negative responsiveness to PTH, calcitonin, and 1 alpha,25(OH)2D3. Biochem Biophys Res Commun 1998; 249 : 11–6. [Google Scholar]
- Murayama A, Takeyama K, Kitanaka S, et al. Positive and negative regulations of the renal 25-hydroxyvitamin D3 1alpha-hydroxylase gene by parathyroid hormone, calcitonin, and 1alpha,25(OH)2D3 in intact animals. Endocrinology 1999; 140 : 2224–31. [Google Scholar]
- Shinki T, Ueno Y, DeLuca HF, Suda T. Calcitonin is a major regulator for the expression of renal 25- hydroxyvitamin D3-1alpha-hydroxylase gene in normocalcemic rats. Proc Natl Acad Sci USA 1999; 96 : 8253–8. [Google Scholar]
- Murayama A, Takeyama K, Asahina T, Kitanaka S, Kato S. Cloning of a novel transcription factor mediating the negative vitamin D responsiveness through the human 25-hydroxyvitamin D-1alpha-hydroxylase nVDRE. J Bone Miner Res 2000; 15 (suppl 1) : S199. [Google Scholar]
- Viville S. Recombinaison homologue : nouveaux vecteurs, nouvelles perspectives. Med Sci 1995; 11 : 735–46. [Google Scholar]
- Dardenne O, Prud’homme J, Arabian A, Glorieux FH, St-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D3-1α-hydroxylase gene (cyp2781) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology 2001; 142 : 3135–41. [Google Scholar]
- Girasole G, Wang JM, Pedrazzoni M, et al. Augmentation of monocyte chemotaxis by 1 alpha,25-dihydroxyvitamin D3. Stimulation of defective migration of AIDS patients. J Immunol 1990; 145 : 2459–64. [Google Scholar]
- Smith EL, Walworth NC, Holick MF. Effect of 1 alpha,25-dihydroxyvitamin D3 on the morphologic and biochemical differentiation of cultured human epidermal keratinocytes grown in serum-free conditions. J Invest Dermatol 1986; 86 : 709–14. [Google Scholar]
- Ducy P, Starbuck M, Priemel M, et al. A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev 1999; 13 : 1025–36. [Google Scholar]
- Kinuta K, Tanaka H, Shinohara M, Kato S, Seino Y. Vitamin D is a negative regulating factor in bone mineralization. J Bone Miner Res 2000;15 (suppl 1) : S180. [Google Scholar]
- Gardiner EM, Sims N, Thomas G, et al. Elevated osteoblastic vitamin D receptor in transgenic mice yields stronger bones. Bone 1998; 23 (suppl 5) : S176. [Google Scholar]
- Tarutani M, Itami S, Okabe M, et al. Tissue-specific knockout of the mouse Pig-a gene reveals important roles for GPI-anchored proteins in skin development. Proc Natl Acad Sci USA 1997; 94 : 7400–5. [Google Scholar]
- Horvai A, Palinski W, Wu H, Moulton KS, Kalla K, Glass CK. Scavenger receptor A gene regulatory elements target gene expression to macrophages and to foam cells of atherosclerotic lesions. Proc Natl Acad Sci USA 1995; 92 : 5391–5. [Google Scholar]
Liste des tableaux
Mutations du gène de la 25-hydroxyvitamine D-1α-hydroxylase chez les patients atteints de rachitisme.
Liste des figures
|
Figure 1. Biosynthèse de la 1,25(OH)2D3. La vitamine D (ici D3 puisque le schéma s’applique à l’homme), synthétisée suite à l’irradiation de la peau par les rayons solaires, est convertie en 25-hydroxyvitamine D3 [25(OH)D3] par la vitamine D 25-hydroxylase au niveau du foie. Ce métabolite inactif est ensuite métabolisé en 1α,25-dihydroxyvitamine D3 [1,25(OH)2D3] à la suite de l’action de l’enzyme rénale, la 25-hydroxyvitamine D-1α-hydroxylase (1α-OHase). L’hormone 1,25(OH)2D3 se lie ensuite à son récepteur nucléaire, le VDR ( vitamin D receptor), un récepteur nucléaire de type II qui forme un dimère actif avec son partenaire, le RXR (retinoid X receptor) , et module la transcription de gènes cibles. Le ligand du RXR, l’acide 9-cisrétinoïque, n’est pas illustré. La cause moléculaire des rachitismes PDDR (pseudo vitamin D deficiency rickets) , – des mutations dans le gène codant pour la 1 α-OHase – est illustrée par la flèche interrompue au niveau du rein. L’autre forme de rachitisme héréditaire vitamino-dépendant, HVD-RR (hereditary vitamin D resistant rickets) , est due à des mutations affectant la structure ou la fonction du VDR, soit au niveau du domaine de liaison du ligand (LBD, ligand binding domain ), soit au niveau du domaine de liaison à l’ADN (DBD, DNA binding domain) . |
| Dans le texte | |
|
Figure 2. Mutations chez les patients atteints de rachitisme PDDR (pseudo-vitamin D deficiency rickets). La structure du gène de la 25-hydroxyvitamine D-1a-hydroxylase (1a-OHase) est représentée de façon schématique. Les exons sont numérotés de 1 à 9; les régions rouges correspondent aux séquences non-traduites en 5’ et en 3’, respectivement. Les 26 mutations identifiées jusquàa présent chez des patients atteints de rachitisme PDDR sont représentées au-dessus et en dessous de la carte du gène. Les nombres indiquent le nucléotide ou l’acide aminé muté, selon le cas. Δ : délétion; IVS3 + 1g->a: mutation dans l’intron 3 (intervening sequence 3) affectant l’epis-sage; 7 bp dupl: duplication de 7 paires de bases; 2 pb dupl: duplication de 2 paires de bases. |
| Dans le texte | |
|
Figure 3. Radiographies du fémur d’animaux déficients pour le gène de la 25-hydroxyvitamine D-1α-hydroxylase (1α-OHase). A. Fémur d’un animal hétérozygote. B. Fémur d’un homozygote mutant pour la 1α-OHase. Notez l’élargissement de la plaque de croissance et la minceur du cortex du fémur mutant, démontrant la présence de rachitisme et d’ostéomalacie. |
| Dans le texte | |
|
Figure 4. Survie des homozygotes mutants pour le gène de la 25-hydroxyvitamine D-1α-hydroxylase (1α-OHase). Les souris déficientes pour le gène 1α-OHase ont é t é sevrées à l’âge de trois semaines et alimentées par un régime normal. Dans ces conditions, les animaux meurent entre la 11e et la 16e semaine de vie. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.