")
")
| Issue |
Med Sci (Paris)
Volume 42, Number 3, Mars 2026
L’antibiorésistance au prisme des trois santés
|
|
|---|---|---|
| Page(s) | 263 - 269 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2026034 | |
| Published online | 20 March 2026 | |
Émergence, évolution et diffusion de l’antibiorésistance
Emergence, evolution and spread of antibiotic resistance
1
AP-HP, Hôpital Bichat-Claude Bernard, Laboratoire de Bactériologie, Paris, France
2
Université Paris Cité et Université Sorbonne Paris Nord, Inserm UMR1137, IAME (infection, antimicrobiens, modélisation, évolution), Paris, France
3
Institut Pasteur, Université de Paris Cité, CNRS, UMR6047, EERA (écologie et l’évolution de la résistance aux antibiotiques), Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
**
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
La résistance aux antibiotiques pose un problème majeur de santé publique, avec plus d’un million de décès qui lui sont attribués chaque année dans le monde. Elle survient chez les bactéries à la suite de mutations ou par le transfert horizontal de gènes de résistance. L’environnement joue un rôle crucial dans l’émergence et la dissémination de ces gènes de résistance, certaines bactéries environnementales servant de réservoir. La résistance aux antibiotiques nécessite ainsi d’être abordée par une approche multisectorielle et multidisciplinaire « One Health » qui englobe les secteurs humain, animal et environnemental. Pour lutter contre l’antibiorésistance, il est essentiel de réduire la consommation d’antibiotiques, d’améliorer les conditions d’hygiène et de renforcer la surveillance.
Abstract
Antibiotic resistance is a major public health issue, responsible for around one million deaths worldwide each year. It arises in bacteria as a result of mutations or horizontal gene transfer of resistance genes. The environment plays a crucial role in the emergence and spread of these genes, with environmental bacteria acting as reservoirs. Addressing antibiotic resistance therefore requires a multisectoral and multidisciplinary “One Health” approach that spans the human, animal and environmental sectors. To combat antimicrobial resistance, it is essential to reduce the use of antibiotic, improve hygiene conditions, and strengthen surveillance.
© 2026 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Inserm).
La résistance aux antibiotiques peut être appréhendée sous différents angles. Sur le plan écologique, la résistance à un antibiotique particulier au sein d’une espèce se manifeste par une sensibilité réduite, comparée à la distribution de la sensibilité observée dans la population naturelle (ou sauvage). D’un point de vue clinique, elle se caractérise par l’absence de réponse d’une bactérie à un antibiotique présent à une concentration non toxique pour le patient. La résistance aux antibiotiques résulte d’un processus évolutif qui conduit à la sélection de bactéries capables de se multiplier en présence d’un inhibiteur de croissance (antibiotique bactériostatique), ou d’une substance mortelle (antibiotique bactéricide). La résistance aux antibiotiques est un processus naturel qui préexistait à leur utilisation en santé humaine et animale [1]. En effet, les antibiotiques sont le plus souvent dérivés de substances naturelles produites par des bactéries ou par des champignons présents dans l’environnement. Par conséquent, les bactéries produisant des antibiotiques ainsi que celles qui y sont potentiellement exposées ont évolué en développant des mécanismes de protection qui peuvent être capturés par les bactéries pathogènes [2]. L’antibiorésistance est un phénomène qui concerne la santé humaine, animale et environnementale et qui épouse ainsi le concept de « une seule santé » (One Health) (Figure 1). De nombreuses espèces bactériennes sont ubiquitaires et présentes au sein de communautés bactériennes complexes. C’est notamment le cas des microbiotes colonisant les humains et les animaux. Les antibiotiques exercent une pression de sélection sur la résistance, y compris sur ces populations commensales, qui constituent des réservoirs de gènes de résistance pour les bactéries pathogènes. De plus, les antibiotiques sont les antimicrobiens les plus consommés en fréquence et en quantité. Ils sont utilisés chez l’être humain, mais encore plus massivement en usage vétérinaire dans les élevages. Ces substances, également excrétées, se retrouvent dans l’environnement. La sélection et la diffusion des bactéries résistantes et des gènes de résistance ont donc lieu dans ces différents secteurs. Face à l’émergence et la diffusion de bactéries résistantes aux antibiotiques, de nouveaux antibiotiques actifs ont été mis sur le marché, en particulier entre les années 1960 et 1980 (période appelée « l’âge d’or » des antibiotiques). Par la suite, la mise sur le marché de nouveaux antibiotiques s’est ralentie, alors que la résistance aux antibiotiques a continué de se développer avec l’apparition de nouveaux gènes de résistance (antibiotic resistance genes [ARG]), définis comme des gènes dont l’expression chez une bactérie diminue sa sensibilité à un antibiotique [3], combinés à la diffusion massive de certains clones bactériens dans les milieux hospitaliers et communautaires. De nombreuses infections bactériennes sont ainsi plus difficiles à traiter, et la mortalité associée à la résistance aux antibiotiques est en croissance, particulièrement dans les pays à revenu faible et intermédiaire [4, 5]. Cette revue a pour objectif d’expliquer les mécanismes par lesquels la résistance aux antibiotiques émerge chez les bactéries pathogènes, ainsi que les déterminants sous-tendant leur diffusion.
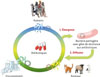 |
Figure 1 Emergence et dissémination de la résistance aux antibiotiques dans les trois secteurs (humain, animal, et environnemental). Le schéma illustre les deux étapes clés du processus. L’émergence correspond à la captation, par des bactéries pathogènes, de gènes de résistance aux antibiotiques provenant de bactéries environnementales ou vivant dans les microbiotes humains ou animaux, favorisée par la présence d’antibiotique. Le cercle représente la diffusion de ces bactéries résistantes entre les humains, les animaux et l’environnement, via des interactions directes ou indirectes telles que l’eau, les sols ou les aliments. Ce cycle met en évidence l’interconnexion entre les différents compartiments et souligne l’importance d’une approche « Une seule santé » (« One Health ») pour limiter la dissémination de la résistance (Figure réalisée avec NIH Bioart (https://bioart.niaid.nih.gov/). |
Acquisition de la résistance par mutation de gènes chromosomiques
Une population bactérienne peut contenir des souches portant des mutations conférant une résistance aux antibiotiques, qui pourront être sélectionnées sous l’action des antibiotiques. Ces souches résistantes pourront recoloniser l’environnement et conduire à une infection insensible à l’antibiotique. Ces mutations ont fréquemment un impact négatif sur le fitness de la souche (c’est-à-dire sur sa capacité à se multiplier dans un environnement donné), limitant ainsi son risque de propagation entre personnes ou dans l’environnement. Néanmoins, dans certains cas, des mutations compensatrices réduisent l’impact négatif des premières mutations sur le fitness. Ces souches résistantes deviennent alors capables de disséminer. La résistance acquise par mutation de gènes chromosomiques est notamment le seul mécanisme connu de résistance chez les bactéries du complexe Mycobacterium tuberculosis, l’agent de la tuberculose. Toutefois, pour d’autres espèces bactériennes, un haut niveau de résistance est fréquemment associé à une combinaison de mutations et à l’acquisition de gènes de résistance aux antibiotiques. Les mutations peuvent conduire à la résistance aux antibiotiques selon des mécanismes divers dépendant de la bactérie et de l’antibiotique.
Les mutations modifiant la cible de l’antibiotique
Les fluoroquinolones ont pour cibles deux topoisomérases1 qui assurent des fonctions essentielles à la survie des bactéries. Des mutations affectant un domaine spécifique de ces protéines les rendent moins sensibles à l’antibiotique sans impacter leur fitness. C’est le mécanisme principal de résistance aux fluoroquinolones pour les entérobactéries comme Escherichia coli ou Klebsiella pneumoniae. Ces molécules sont associées à l’émergence de certains clones bactériens résistants, comme le clone ST131-C de E. coli ou ST258 de K. pneumoniae. Les mutations de la protéine cible peuvent précéder l’acquisition de gènes leur conférant des résistances de plus haut niveau. C’est par exemple le cas chez E. coli, où des combinaisons de mutations sont apparues dans le gène codant la protéine liant la pénicilline 3 (PLP3) et qui est impliquée dans la biosynthèse de la paroi bactérienne. Ces mutations réduisent la sensibilité aux bêta-lactamines, telles que l’aztréonam ou la ceftazidime, qui ciblent principalement cette PLP. Il a été montré que ces mutations pouvaient précéder l’acquisition de gènes de carbapénèmase (codant des enzymes dégradant les carbapénèmes, des bêta-lactamines de dernier recours) et qu’elles avaient contribué à l’émergence et à la dissémination de clones résistants aux antibiotiques de dernière génération, tel que le céfidérocol [6].
La recombinaison et l’échange de gènes contribuent également à l’acquisition de variants insensibles à l’antibiotique. C’est le cas des combinaisons de mutations dans PLP3 de E. coli qui n’ont probablement eu lieu qu’une seule fois, mais qui ont disséminé dans de nombreux lignages par transfert génétique horizontal et recombinaison. Chez Streptococcus pneumoniae, la situation est plus complexe, avec l’apparition de mutations dans les gènes de PLP d’espèces commensales des voies aériennes supérieures comme Streptococcus mitis et Streptococcus oralis, phylogénétiquement proches du pneumocoque, suivies d’événements de recombinaison aboutissant à des gènes mosaïques produisant des protéines à sensibilité réduite aux bêtalactamines (« PLP mosaïques »). De manière intéressante, les vaccins multivalents contre les infections invasives à pneumocoque (comme le Prevenar-20) ciblent des sérotypes associés à ces clones résistants à la pénicilline et ont contribué à une diminution de la résistance à cet antibiotique chez cette bactérie [7].
Les mutations affectant la perméabilité de la paroi
Chez les bactéries à Gram négatif, la membrane externe constitue une barrière à l’entrée de petites molécules hydrophiles, dont de nombreux antibiotiques, notamment les bêta-lactamines et certaines fluoroquinolones. Ces molécules passent au travers de pores constitués de protéines appelées porines. L’inactivation des gènes de porines réduit l’entrée des antibiotiques et de certains nutriments, pouvant induire un coût de fitness, comme chez K. pneumoniae avec la modification de la porine OmpK-36. On trouve fréquemment, dans les souches résistantes aux carbapénèmes, comme le clone ST512 exprimant la carbapénèmase KPC, une insertion dans une boucle de la porine OmpK-36, réduisant la taille du canal et, par conséquent, l’entrée des bêta-lactamines, avec un impact sur le fitness plus faible qu’en l’absence totale de cette porine [8]. Chez Pseudomonas aeruginosa, l’inactivation de la porine D2 est fréquemment retrouvée dans les souches résistantes à l’imipénème (un carbapénème largement prescrit en France et constituant l’une des dernières options de traitement).
Les mutations augmentant l’expression des gènes de résistance
De nombreuses bactéries portent des gènes chromosomiques de résistance aux antibiotiques, partagés par l’ensemble des isolats d’une espèce, comme des bêta-lactamases ou des pompes d’efflux. Ces gènes sont fréquemment exprimés à un faible niveau, avec un effet limité sur la sensibilité aux antibiotiques. Chez E. coli, des mutations dans la région promotrice du gène ampC codant une céphalosporinase augmentent son expression [9]. Chez P. aeruginosa comme chez les entérobactéries du groupe 3 (par exemple les genres Enterobacter et Serratia), l’expression du gène codant pour la céphalosporinase AmpC est soumise à une régulation complexe, et des mutations dans certains composants de cette régulation entraînent une surexpression et une résistance aux pénicillines et aux céphalosporines de 3e génération. La combinaison de la surexpression de ampC, de pompes d’efflux et de l’inactivation du gène oprD se traduit par une résistance à de nombreuses bêta-lactamines sans acquisition de gène codant une bêta-lactamase.
Acquisition et évolution des gènes de résistance aux antibiotiques
Émergence des gènes de résistance aux antibiotiques chez les bactéries pathogènes
L’émergence de la résistance par acquisition de gènes de résistance aux antibiotiques soulève la question de leur origine. Ce phénomène résulte souvent de la capture de ces gènes à partir de bactéries environnementales ou vivant dans des microbiotes humains ou animaux, suivie de leur adaptation et de leur évolution. Les gènes de résistance aux antibiotiques existaient en effet bien avant la consommation à grande échelle de ces molécules [1]. Ces gènes peuvent conférer une activité contre une large gamme d’antibiotiques, y compris – malheureusement – contre les nouvelles molécules récemment mises sur le marché [10]. Il est donc fondamental de caractériser les phénomènes d’émergence de la résistance, d’en retracer l’origine et d’élucider les mécanismes moléculaires et écologiques impliqués dans leur capture. Les gènes de résistance aux antibiotiques sont fréquemment associés à des éléments génétiques mobiles, tels que les plasmides, les transposons conjugatifs et, plus rarement, les bactériophages. La colocalisation de plusieurs gènes de résistance aux antibiotiques sur un même élément génétique mobile favorise leur transfert simultané entre bactéries, facilitant ainsi l’émergence de phénotypes multirésistants. Ce regroupement génétique est à l’origine de phénomènes de cosélection, dans lesquels l’exposition à un seul antibiotique peut exercer une pression de sélection sur l’ensemble des résistances portées par l’élément génétique mobile, y compris contre des molécules non administrées. Il a également été proposé que l’exposition aux antiseptiques peut co-sélectionner des résistances aux antibiotiques [11]. À ce jour, plusieurs milliers de gènes de résistance aux antibiotiques ont été identifiés et répertoriés dans des bases de données spécialisées, telles que la comprehensive antibiotic resistance database (CARD2, [12]) ou ResFinder3 [13]. Chez les bactéries pathogènes pour l’être humain et les animaux (bactéries pathogènes strictes et pathogènes opportunistes), l’origine des gènes de résistance aux antibiotiques acquis reste le plus souvent mal connue, ce qui rend difficile la reconstitution de l’événement d’émergence [14]. Le scénario le plus probable est la capture d’un gène chromosomique d’une bactérie environnementale vers un élément génétique mobile par l’intermédiaire d’éléments génétiques intégratifs comme des séquences d’insertion (IS) et les transposons, ou par les intégrons, plateforme de mobilité des gènes de résistance. Cet élément génétique mobile peut ensuite être transmis, directement ou indirectement, aux bactéries pathogènes. Dans toutes ces étapes, la pression de sélection exercée par les antibiotiques joue un rôle important. La caractérisation du gène de résistance à la colistine, mcr (mobile colistin resistance), est en cela exceptionnelle puisque tous les éléments concourant à l’émergence de ce gène chez E. coli ont pu être identifiés : la bactérie donneuse (une bactérie du genre Moraxella), le véhicule (la séquence d’insertion ISApl1), l’hôte (le porc) et le contexte (élevage porcin et utilisation massive de la colistine comme complément alimentaire et facteur de croissance utilisé dans les élevages de certains pays) [15].
Lorsqu’un nouvel antibiotique appartenant à une famille déjà utilisée est mis sur le marché, des souches résistantes émergent rapidement. Ce nouvel antibiotique peut en effet sélectionner des variants de gènes de résistance aux antibiotiques agissant contre des membres de cette famille déjà prescrits, qui deviennent actifs contre ce nouvel antibiotique, à la suite de mutations ou directement via l’émergence de nouveaux gènes de résistance aux antibiotiques. L’utilisation de céphalosporines à large spectre a entraîné l’émergence de bêta-lactamases à spectre étendu (BLSE) dérivées des bêta-lactamases à spectre étroit, TEM et SHV, qui, suite à des mutations, étaient devenues capables d’hydrolyser les céphalosporines à large spectre, mais également à celle de nouveaux gènes de résistance aux antibiotiques comme ceux codant pour les protéines CTX-M. Ces derniers gènes ont été capturés de bactéries du genre Kluyvera et ont disséminé parmi les entérobactéries après leur transfert sur des plasmides conjugatifs. Un variant, blaCTX-M-15, a évolué à partir du gène capturé pour acquérir une activité plus importante contre la ceftazidime, une céphalosporine de 3e génération, à la suite d’une seule substitution, Asp240Gly. Le lignage de E. coli ST131-C2 a contribué à la dissémination de ce variant [16]. La combinaison ceftazidime-avibactam a été introduite récemment pour le traitement des infections à entérobactéries productrices de carbapénèmases, notamment les enzymes de classe A comme KPC, mais elle est inefficace contre les métallo-β-lactamases (classe B), telles que NDM et VIM. Des cas de résistance ont été rapportés chez des souches exprimant des variantes mutées de KPC, souvent localisées dans la boucle oméga4 (Ω-loop) de l’enzyme. Ces mutations augmentent l’affinité de KPC pour la ceftazidime, réduisant l’efficacité de l’inhibition par l’avibactam et entraînant un échec thérapeutique. Paradoxalement, ces souches mutées présentent une activité carbapénémase affaiblie, ce qui peut restaurer leur sensibilité aux carbapénèmes [17]. Ces observations illustrent les limites des nouvelles associations antibiotiques face à l’émergence rapide de mécanismes de résistance adaptatifs.
Rôle de l’environnement
L’environnement apparaît comme le réservoir principal des gènes de résistance aux antibiotiques pour les bacilles à Gram négatif du phylum des Pseudomonadota (qui comprend les entérobactéries et les bacilles non fermentaires comme P. aeruginosa et Acinetobacter baumannii) [18]. Par exemple, des bactéries du genre Shewanella vivant dans des environnements aquatiques ont été identifiées comme les hôtes à partir desquels les gènes blaOXA-48 (résistance aux carbapénèmes), qnrA (résistance aux fluoroquinolones) et mcr-4 (résistance à la colistine) ont été transférés vers des bacilles à Gram négatif pathogènes comme les entérobactéries [19–21]. Les eaux usées ont ainsi été identifiées comme un point d’émergence de gènes de résistance aux antibiotiques provenant de bactéries environnementales et transférés vers des bactéries pathogènes. En effet, les eaux usées offrent des conditions favorables en mettant en présence des bactéries hydriques, comme Shewanella, des bactéries pathogènes opportunistes de notre microbiote intestinal (entérobactéries, entérocoques), ainsi que des éléments génétiques mobiles à l’origine de la mobilisation de gènes de résistance aux antibiotiques [22].
Rôle des microbiotes
Les microbiotes humains, ainsi que ceux des animaux sont également suspectés d’être des réservoirs de nouveaux gènes de résistance aux antibiotiques pour les bactéries pathogènes. Plusieurs études, basées soit sur la recherche systématique de gènes de résistance par expression chez E. coli (métagénomique fonctionnelle), soit par des analyses bio-informatique de métagénomes, et la confirmation expérimentale (par synthèse de gène et clonage) de la capacité d’un gène à rendre une bactérie résistante, ont confirmé que les bactéries composant ces microbiotes, notamment intestinaux, étaient porteuses d’une grande diversité de gènes de résistance aux antibiotiques [23–25]. Toutefois, plusieurs éléments vont à l’encontre du partage de ces gènes de résistance aux antibiotiques entre les bactéries commensales et les bactéries pathogènes. Les gènes de résistance aux antibiotiques des bactéries commensales présentent pour la plupart une séquence nucléotidique très différente de celle des gènes de résistance aux antibiotiques trouvés chez les bactéries pathogènes [23]. La majorité de ces gènes de résistance aux antibiotiques sont intégrés dans le chromosome de leur hôte sans être associés, dans leur environnement, à des gènes de mobilité [23]. Il semble aussi que des barrières phylogénétiques restreignent l’émergence des gènes de résistance aux antibiotiques, puisqu’on observe très rarement une acquisition depuis un phylum différent de celui de la bactérie réceptrice [26]. Pour les entérobactéries, le gène tet(X) et le gène blaMUN-1 (tous deux retrouvés chez des bactéries du phylum Bacteroidota) font figure d’exception. Chez les Bacillota (ex-Firmicutes), toutefois, le partage de gènes de résistance aux antibiotiques dans le microbiote intestinal est bien décrit, par exemple pour le gène vanB présent chez Enterococcus spp. et différentes espèces du genre Clostridium [27].
Diffusion de l’antibiorésistance
L’analyse de la propagation de la résistance aux antibiotiques s’appuie sur des données de surveillance de l’antibiorésistance à différents niveaux géographiques et pendant plusieurs années. Ces données comprennent des informations épidémiologiques sur la résistance de diverses bactéries pathogènes à différents antibiotiques, ainsi que des données moléculaires sur les souches et les gènes de résistance aux antibiotiques.
La dissémination de la résistance aux antibiotiques soulève des questions cruciales concernant les mécanismes et les voies de transmission, ainsi que les facteurs favorisant ce phénomène, sans oublier les obstacles potentiels. Ces informations sont indispensables pour prévenir ou limiter l’émergence et la dissémination de la résistance. L’avènement du séquençage à haut débit et la réduction de son coût ont transformé l’épidémiologie moléculaire de la résistance aux antibiotiques, permettant de suivre la dissémination des clones et plasmides multirésistants et, dans certains cas, de reconstituer des histoires complexes. L’étude de l’émergence et de la propagation de la résistance aux antibiotiques est intrinsèquement multidisciplinaire, combinant la surveillance, des études épidémiologiques, écologiques et sociologiques, des analyses génomiques et évolutives, des investigations moléculaires et mécanistiques, ainsi que des outils de modélisation mathématique. L’objectif est d’intégrer ces diverses données pour élaborer des stratégies de prévention efficaces.
Une seule santé : diffusion dans les secteurs humain, animal et environnemental
Le concept « une seule santé » (“One Health”) reconnaît que la santé des secteurs humain, animal et environnemental est interconnectée. Il s’applique tout à fait à l’antibiorésistance en cela que les bactéries résistantes et les éléments génétiques mobiles porteurs de gènes de résistance aux antibiotiques circulent dans ces trois secteurs [28] (→).
(→) Voir m/s n° 3, 2025, page 211
De même, les antibiotiques sont largement consommés en médecine vétérinaire et en agriculture (représentant environ 70 % de la consommation mondiale d’antibiotiques), pour traiter les infections, les prévenir à l’échelle d’un élevage (métaphylaxie), mais aussi comme facteurs de croissance des animaux de rente [29]. Cette pratique est d’ailleurs interdite en Europe depuis 2006, mais continue d’être utilisée dans de nombreux pays, tels que les États-Unis et la Chine. Les antibiotiques, quelle que soit leur indication, peuvent se retrouver dans les eaux usées, notamment pour les plus stables d’entre eux (par exemple la tétracycline ou les fluoroquinolones) [30].
Les bactéries multirésistantes peuvent ainsi circuler dans les trois secteurs. L’Organisation mondiale de la santé (OMS) a proposé un protocole de surveillance « One Health » (appelé « Tricycle ») simple et standardisé, consistant à mesurer la prévalence des E. coli producteurs de bêta-lactamases à spectre étendu (BLSE) chez les sujets communautaires (femmes enceintes), dans les bactériémies5, dans les eaux usées et dans les poulets (vendus sur les marchés) [31] (→).
(→) Voir m/s n° 3, 2025, page 222
Une étude basée sur ce protocole menée à Madagascar a montré le partage entre les différents compartiments de E. coli, mais aussi des plasmides portant les gènes codant les BLSE [32]. Toutefois, ce partage semble bien plus rares dans d’autres contextes comme au Royaume-Uni [33].
Rôle de la globalisation
L’augmentation de la prévalence du portage intestinal de bactéries multirésistantes (particulièrement des entérobactéries) a très tôt fait craindre que l’exposition au péril fécal durant les voyages joue un rôle majeur dans la diffusion de la résistance aux antibiotiques. Dès 1990, Murray et al. ont rapporté l’acquisition de E. coli multirésistants chez des individus en bonne santé n’ayant pas pris d’antibiotiques après un voyage au Mexique [34]. La diffusion des entérobactéries productrices de bêta-lactamases à spectre étendu dans la communauté, ainsi que l’augmentation du tourisme international (543 millions en 1995 contre 1,76 milliard en 2019) ont conduit à la réalisation d’études mesurant le taux d’acquisition de bactéries multirésistantes par les voyageurs [35, 36]. Ces études ont montré qu’une proportion importante d’entre eux (21 à 51 %) devenaient porteurs d’entérobactéries multirésistantes durant un séjour à l’étranger [37]. Les facteurs de risque associés à cette acquisition étaient le séjour dans certaines régions (sous-continent indien et Asie du Sud-Est), la prise d’antibiotiques durant le voyage, le fait d’effectuer un séjour hors des hôtels-clubs fermés, et la survenue de troubles digestifs durant le séjour. Les entérobactéries acquises durant le séjour étaient majoritairement des E. coli produisant des bêta-lactamases à spectre étendu de type CTX-M. La majorité des voyageurs n’hébergeait plus d’entérobactéries multirésistantes un mois après la fin du voyage [37]. Cependant, certains sujets pouvaient encore être porteurs 12 mois après le retour. Le portage prolongé au retour de voyage semble multifactoriel, les souches persistantes étant des E. coli appartenant aux phylogroupes B2 et D et présentes en concentrations plus importantes [38, 39]. De plus, les sujets présentant un portage persistant un mois après le retour avaient une composition du microbiote intestinal différente de celle des sujets décolonisés, avec notamment une abondance moindre de bactéries du genre Bifidobacterium [40].
Contribution de la modélisation pour identifier les facteurs de l’antibiorésistance
La résistance aux antibiotiques est globalement plus élevée dans les pays à revenu faible ou intermédiaire que dans les pays à revenu élevé [5]. La mortalité associée ou attribuable à l’antibiorésistance y est également la plus élevée, notamment en raison de la difficulté d’accès aux antibiotiques actifs contre les bactéries multirésistantes [4]. La résistance aux antibiotiques d’un pays n’est pas seulement corrélée à sa consommation d’antibiotiques. En effet, particulièrement pour les bactéries entériques (comme les entérobactéries), la diffusion de la résistance est facilitée par le manque d’infrastructures d’assainissement des eaux usées et plus globalement, par la difficulté d’accéder à une eau et à des aliments non contaminés par des matières fécales humaines ou animales [41, 42]. L’intégration de ces données dans des modèles mathématiques permet de déterminer les facteurs qui influencent les niveaux de résistance observés dans différents pays et leur propagation. Une analyse des données de résistance présentes dans la base de données ATLAS6 pour 13 couples bactérie/antibiotique dans 51 pays, entre 2006 et 2019, a permis de montrer que le facteur principal contribuant au niveau de résistance était la qualité du système de santé alors que la consommation d’antibiotiques n’intervenait que pour les E. coli et P. aeruginosa résistants aux fluoroquinolones, et A. baumannii résistant aux carbapénèmes [43].
Conclusion
L’antibiorésistance est un phénomène naturel par lequel des mécanismes permettent aux bactéries de survivre face aux traitements antibiotiques. L’immense diversité génétique chez les bactéries soutient l’idée que des mécanismes de résistance peuvent préexister à tout nouvel antibiotique, et que leur émergence chez les bactéries pathogènes est inévitable. L’émergence de la résistance est influencée par la consommation d’antibiotiques en médecine humaine, vétérinaire et en agriculture, s’inscrivant dans le concept « One Health ». Les antibiotiques favorisent la sélection de gènes de résistance présents dans ces écosystèmes et leur transfert potentiel vers les bactéries pathogènes, comme cela a été montré pour mcr-1. Au-delà de l’émergence de la résistance aux antibiotiques, la diffusion se fait par transmission horizontale ou verticale, facilitée par la globalisation et les voyages. La résistance est plus élevée dans les pays à faibles revenus et dans les pays tropicaux en raison notamment de l’accès limité aux infrastructures d’assainissement et à une consommation d’antibiotiques non contrôlée.
Cet état des lieux appelle à la mise en place de mesures visant à contenir l’antibiorésistance et à prévenir l’émergence de nouvelles menaces. Améliorer nos capacités de surveillance de la résistance, comme le permet aujourd’hui la plateforme ABRomics7, doit permettre de suivre avec précision l’évolution de la résistance chez les bactéries pathogènes et dans les écosystèmes, et d’anticiper les risques. Plusieurs leviers doivent aussi être actionnés, parmi lesquels réduire la consommation d’antibiotiques, promouvoir le concept d’antibiotiques à faible impact sur les microbiotes humains et animaux, mais aussi à faible impact environnemental (antibiotiques à dégradation rapide) et améliorer les conditions d’hygiène de vie et d’hygiène hospitalière dans les pays à revenu faible et intermédiaire. Ces actions doivent être considérées comme prioritaires pour la santé globale.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article
Références
- D’Costa VM, King CE, Kalan L, et al. Antibiotic resistance is ancient. Nature 2011 ; 477 : 457–61. [Google Scholar]
- Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev 2010 ; 74 : 417–33. [Google Scholar]
- Martínez JL, Coque TM, Baquero F. What is a resistance gene? Ranking risk in resistomes. Nat Rev Microbiol 2015 ; 13 : 116–23. [Google Scholar]
- Naghavi M, Vollset SE, Ikuta KS, et al. Global burden of bacterial antimicrobial resistance 1990–2021: a systematic analysis with forecasts to 2050. Lancet 2024 ; 404 : 1199–226. [Google Scholar]
- Murray CJ, Ikuta KS, Sharara F, et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022 ; 399 : 629–55. [CrossRef] [PubMed] [Google Scholar]
- Patiño-Navarrete R, Rosinski-Chupin I, Cabanel N, et al. Stepwise evolution and convergent recombination underlie the global dissemination of carbapenemase-producing Escherichia coli. Genome Med 2020 ; 12 : 10. [Google Scholar]
- Dagan R, Klugman KP. Impact of conjugate pneumococcal vaccines on antibiotic resistance. Lancet Infect Dis 2008 ; 8 : 785–95. [Google Scholar]
- Wong JLC, Romano M, Kerry LE, et al. OmpK36-mediated Carbapenem resistance attenuates ST258 Klebsiella pneumoniae in vivo. Nat Commun 2019 ; 10 : 3957. [Google Scholar]
- Jaurin B, Grundström T, Normark S. Sequence elements determining ampC promoter strength in E. coli. EMBO J 1982 ; 1 : 875–81. [Google Scholar]
- Gschwind R, Bonnet M, Abramova A, et al. Cefiderocol resistance genes identified in environmental samples using functional metagenomics. ISME J 2026 ; wrag010. [Google Scholar]
- Basiry D, Entezari Heravi N, Uluseker C, et al. The effect of disinfectants and antiseptics on co- and cross-selection of resistance to antibiotics in aquatic environments and wastewater treatment plants. Front Microbiol 2022 ; 13 : 1050558. [Google Scholar]
- Alcock BP, Huynh W, Chalil R, et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res 2023 ; 51 : D690–9. [Google Scholar]
- Bortolaia V, Kaas RS, Ruppe E, et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother 2020 ; 75 : 3491–500. [Google Scholar]
- Ebmeyer S, Kristiansson E, Larsson DGJ. A framework for identifying the recent origins of mobile antibiotic resistance genes. Commun Biol 2021 ; 4 : 1–10. [CrossRef] [PubMed] [Google Scholar]
- Liu Y-Y, Wang Y, Walsh TR, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis 2016 ; 16 : 161–8. [CrossRef] [PubMed] [Google Scholar]
- D’Andrea MM, Arena F, Pallecchi L, et al. CTX-M-type β-lactamases: a successful story of antibiotic resistance. Int J Med Microbiol 2013 ; 303 : 305–17. [Google Scholar]
- Oueslati S, Iorga BI, Tlili L, et al. Unravelling ceftazidime/avibactam resistance of KPC-28, a KPC-2 variant lacking carbapenemase activity. J Antimicrob Chemother 2019 ; 74 : 2239–46. [Google Scholar]
- Forsberg KJ, Patel S, Gibson MK, et al. Bacterial phylogeny structures soil resistomes across habitats. Nature 2014 ; 509 : 612–6. [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Zhang H, Hou M, Xu Y, et al. Action and mechanism of the colistin resistance enzyme MCR-4. Commun Biol 2019 ; 2 : 36. [Google Scholar]
- Poirel L, Rodriguez-Martinez J-M, Mammeri H, et al. Origin of plasmid-mediated quinolone resistance determinant QnrA. Antimicrob Agents Chemother 2005 ; 49 : 3523–5. [Google Scholar]
- Tacão M, Araújo S, Vendas M, et al. Shewanella species as the origin of blaOXA-48 genes: insights into gene diversity, associated phenotypes and possible transfer mechanisms. Int J Antimicrob Agents 2018 ; 51 : 340–8. [Google Scholar]
- Berglund F, Ebmeyer S, Kristiansson E, et al. Evidence for wastewaters as environments where mobile antibiotic resistance genes emerge. Commun Biol 2023 ; 6 : 321. [Google Scholar]
- Ruppé E, Ghozlane A, Tap J, et al. Prediction of the intestinal resistome by a three-dimensional structure-based method. Nat Microbiol 2019 ; 4 : 112–23. [Google Scholar]
- Sommer MOA, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 2009 ; 325 : 1128–31. [Google Scholar]
- Berglund F, Marathe NP, Österlund T, et al. Identification of 76 novel B1 metallo-β-lactamases through large-scale screening of genomic and metagenomic data. Microbiome 2017 ; 5 : 134. [Google Scholar]
- Lund D, Parras-Moltó M, Inda-Díaz JS, et al. Genetic compatibility and ecological connectivity drive the dissemination of antibiotic resistance genes. Nat Commun 2025 ; 16 : 2595. [Google Scholar]
- Ballard SA, Grabsch EA, Johnson PDR, et al. Comparison of three PCR primer sets for identification of vanb gene carriage in feces and correlation with carriage of vancomycin-resistant enterococci: interference by vanB-containing anaerobic bacilli. Antimicrob Agents Chemother 2005 ; 49 : 77–81. [Google Scholar]
- Lacotte Y, Jouvin-Marche E, Ploy MC. EU-JAMRAI – L’Europe engagée sur une réponse coordonnée et « une seule santé » pour faire face à l’antibiorésistance. Med Sci (Paris) 2025 ; 41 : 211–2. [Google Scholar]
- Van Boeckel TP, Glennon EE, Chen D, et al. Reducing antimicrobial use in food animals. Science 2017 ; 357 : 1350–2. [NASA ADS] [CrossRef] [PubMed] [Google Scholar]
- Larsson DGJ, Pedro C de, Paxeus N. Effluent from drug manufactures contains extremely high levels of pharmaceuticals. J Hazard Mater 2007 ; 148 : 751–5. [Google Scholar]
- Milenkov M, Armand-Lefevre L. Surveillance Tricycle-OMS à Antananarivo (Madagascar) - Circulation de souches de Escherichia coli productrices de β-lactamase à spectre étendu et des plasmides entre l’Homme, les poulets et l’environnement. Med Sci (Paris) 2025 ; 41 : 222–6. [Google Scholar]
- Milenkov M, Proux C, Rasolofoarison TL, et al. Implementation of the WHO Tricycle protocol for surveillance of extended-spectrum β-lactamase producing Escherichia coli in humans, chickens, and the environment in Madagascar: a prospective genomic epidemiology study. Lancet Microbe 2024 ; 5 : 100850. [Google Scholar]
- Ludden C, Raven KE, Jamrozy D, et al. One Health Genomic Surveillance of Escherichia coli Demonstrates Distinct Lineages and Mobile Genetic Elements in Isolates from Humans versus Livestock. mBio 2019 ; 10 : e02693–18. [Google Scholar]
- Murray BE, Mathewson JJ, DuPont HL, et al. Emergence of resistant fecal Escherichia coli in travelers not taking prophylactic antimicrobial agents. Antimicrob Agents Chemother 1990 ; 34 : 515–8. [Google Scholar]
- Ruppé E, Armand-Lefèvre L, Estellat C, et al. High rate of acquisition but short duration of carriage of multidrug-resistant enterobacteriaceae after travel to the tropics. Clin Infect Dis 2015 ; 61 : 593–600. [Google Scholar]
- Arcilla MS, Hattem JM van, Haverkate MR, et al. Import and spread of extended-spectrum β-lactamase-producing Enterobacteriaceae by international travellers (COMBAT study): a prospective, multicentre cohort study. Lancet Infect Dis 2017 ; 78–85. [Google Scholar]
- Ruppé E, Andremont A, Armand-Lefèvre L. Digestive tract colonization by multidrug-resistant Enterobacteriaceae in travellers: An update. Travel Med Infect Dis 2018 ; 21 : 28–35. [Google Scholar]
- Cotto O, Armand-Lefèvre L, Matheron S, et al. Within-host dynamics and duration of colonization of travel-acquired multidrug-resistant enterobacterales in untreated individuals. Clin Infect Dis 2023 ; 77 : 934–6. [Google Scholar]
- Armand-Lefèvre L, Rondinaud E, Desvillechabrol D, et al. Dynamics of extended-spectrum beta-lactamase-producing Enterobacterales colonization in long-term carriers following travel abroad. Microb Genom 2021 ; 7 : 000576. [Google Scholar]
- Leo S, Lazarevic V, Gaïa N, et al. The intestinal microbiota predisposes to traveler’s diarrhea and to the carriage of multidrug-resistant Enterobacteriaceae after traveling to tropical regions. Gut Microbes 2019 ; 1–11. [Google Scholar]
- Li W, Huang T, Liu C, et al. Changing climate and socioeconomic factors contribute to global antimicrobial resistance. Nat Med 2025 ; 1–11. [Google Scholar]
- Collignon P, Beggs JJ, Walsh TR, et al. Anthropological and socioeconomic factors contributing to global antimicrobial resistance: a univariate and multivariable analysis. Lancet Planet Health 2018 ; 2 : e398–e405. [CrossRef] [PubMed] [Google Scholar]
- Rahbe E, Watier L, Guillemot D, et al. Determinants of worldwide antibiotic resistance dynamics across drug-bacterium pairs: a multivariable spatial-temporal analysis using ATLAS. Lancet Planet Health 2023 ; 7 : e547–57. [Google Scholar]
Les ADN-topoisomérases sont des enzymes qui contrôlent la structure topologique de l’ADN en générant des coupures transitoires (ndlr).
La boucle oméga est un motif structurel protéique non régulier, constitué d’une boucle de six résidus d’acides aminés ou plus et de toute séquence d’acides aminés qui adopte une conformation spécifique sans appartenir à une structure secondaire régulière comme l’hélice α ou le feuillet β.
La bactériémie est définie par la présence d’une bactérie pathogène dans le sang circulant, authentifiée par des hémocultures positives. Cette présence peut être éphémère ou chronique et peut être accompagnée de signes cliniques ou non (ndlr).
Liste des figures
|
Figure 1 Emergence et dissémination de la résistance aux antibiotiques dans les trois secteurs (humain, animal, et environnemental). Le schéma illustre les deux étapes clés du processus. L’émergence correspond à la captation, par des bactéries pathogènes, de gènes de résistance aux antibiotiques provenant de bactéries environnementales ou vivant dans les microbiotes humains ou animaux, favorisée par la présence d’antibiotique. Le cercle représente la diffusion de ces bactéries résistantes entre les humains, les animaux et l’environnement, via des interactions directes ou indirectes telles que l’eau, les sols ou les aliments. Ce cycle met en évidence l’interconnexion entre les différents compartiments et souligne l’importance d’une approche « Une seule santé » (« One Health ») pour limiter la dissémination de la résistance (Figure réalisée avec NIH Bioart (https://bioart.niaid.nih.gov/). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.