")
")
| Issue |
Med Sci (Paris)
Volume 40, Novembre 2024
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 40 - 44 | |
| Section | Prix SFM | |
| DOI | https://doi.org/10.1051/medsci/2024129 | |
| Published online | 18 November 2024 | |
Altérations métaboliques dans la dystrophie myotonique de type I
Une piste thérapeutique potentielle
Metabolic dysfunctions in type I myotonic dystrophy: A potential therapeutic target
1
Institut NeuroMyoGène, Laboratoire Physiopathologie et Génétique du Neurone et du Muscle, CNRS UMR 5261 - Inserm U1315, Université Claude Bernard Lyon 1, Lyon, France
2
Département des pathologies neuromusculaires, Hôpital Neurologique Pierre Wertheimer, Hospices Civils de Lyon, France
3
Département de Médecine interne, Hôpital Edouard Herriot, Hospices Civils de Lyon, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
La dystrophie myotonique de type I (DM1) est une maladie génétique responsable d’une altération multi-systémique de l’épissage alternatif. En conséquence, de nombreuses voies de signalisation cellulaires sont dérégulées. Une répression de l’AMPK (adenosine monophosphate activated kinase), le principal régulateur du métabolisme cellulaire, est notamment observée. Restaurer la voie de signalisation de l’AMPK pourrait permettre d’améliorer la biogenèse et la dynamique mitochondriales, les processus mitophagiques et de régulation du stress oxydatif mitochondrial, la production énergétique et, in fine, l’homéostasie du tissu musculaire strié squelettique dans la DM1.
Abstract
Myotonic dystrophy type I (DM1) is a genetic disease characterized by a multisystemic RNA metabolism dysregulation and splicing toxicity. Numerous signaling pathways are deregulated, and especially AMPK, a key sensor and regulator of cellular metabolism. To restore AMPK signaling activity in DM1 muscle tissue and cells could improve mitochondrial biogenesis and dynamics, mitophagy and oxidative stress, energy production and, in fine, skeletal muscle tissue homeostasis.
© 2024 médecine/sciences – Inserm

© L. Lessard
Vignette : Le complexe AMPK, une cible thérapeutique dans la DM1. (Créée avec BioRender.com)
La dystrophie myotonique de type 1 (DM1) est la dystrophie musculaire la plus fréquente en population adulte [1]. Il s’agit d’une maladie autosomique dominante dont le spectre clinique est extrêmement varié. La forme débutant à l’âge adulte se présente classiquement par une myotonie et un déficit moteur des muscles distaux, s’associant de manière variable à des atteintes respiratoires, cardiaques, endocriniennes, ophtalmologiques et neurologiques centrales [2, 3]. Il n’existe pas de traitement curatif.
La DM1 est causée par une expansion de triplets CTG, située dans la partie non codante 3’ du gène DMPK (dystrophia myotonica protein kinase). Les individus sains possèdent de 5 à 37 répétitions, lorsque les patients atteints de DM1 en ont de 50 à plus de 3 000. Cette expansion de CTG est instable sur le plan somatique et sur le plan germinal. L’augmentation du nombre de répétitions au cours de la gamétogenèse chez le parent affecté est, dans une certaine mesure, corrélée avec l’augmentation de la sévérité clinique et la précocité de la maladie chez l’enfant atteint (phénomène d’anticipation) [3]. Les transcrits DMPK porteurs de l’expansion CUG (CUGexp) forment des structures anormales en « épingles à cheveux » qui s’agrègent en foci nucléaires. Ces foci altèrent la localisation et la fonction de plusieurs protéines de liaison à l’ARN, ce qui altère les mécanismes de l’épissage de centaines de transcrits [4]. De très nombreuses fonctions cellulaires et tissulaires sont perturbées par ces altérations de l’épissage : des processus de régulation du cycle cellulaire et de la sénescence, ainsi que des mécanismes autophagiques, apoptotiques, inflammatoires, du système ubiquitine-protéasome, de l’équilibre synthèse/dégradation protéique, de régulation des micro-ARN, de sensibilité à l’insuline, d’homéostasie calcique, etc. Plusieurs gènes dont l’épissage est altéré peuvent être reliés aux principales caractéristiques phénotypiques de la DM1 : SCN5A (canal sodique Nav1.5) et la cardiopathie, INSR (récepteur à l’insuline) et l’insulinorésistance/diabète, SYN1 (synapsine 1, vésicules synaptiques) et GLT1 (transporteur du glutamate) et l’atteinte cognitive, CLCN1 (canal chlore) et la myotonie. Enfin, sont aussi impactés des gènes clés de la structure et de la fonction du muscle squelettique strié tels que DMD (dystrophine), BIN1 (bridging integrator 1), TNNT3 (troponine T3), TTN (titine), RYR1 (récepteur à la ryanodine 1), etc. [4]
Altérations métaboliques cliniques et leurs conséquences sur le muscle strié squelettique
Parmi les nombreux organes et systèmes affectés dans la DM1, l’altération du métabolisme du glucose, qui peut se manifester par une intolérance au glucose, voire un diabète, représente une cause de morbidité non négligeable. Par ailleurs, d’autres dysfonctions endocriniennes peuvent être observées (fonctions thyroïdienne et parathyroïdienne, hypogonadisme avec insuffisance androgénique et atteinte de l’axe corticotrope) [5] et sont susceptibles d’affecter l’homéostasie du muscle strié squelettique. Or, les dysfonctions endocriniennes et les altérations musculaires sont interdépendantes. En effet, si les signaux métaboliques systémiques (hormones, facteurs de croissance, cytokines, etc.) représentent un niveau de régulation important de l’homéostasie du muscle strié squelettique, ce dernier possède également ses propres fonctions endocrines. Chez un adulte sain, le tissu musculaire squelettique représente environ 45 % du poids du corps total, et c’est un déterminant majeur du métabolisme corporel [6]. Autrement dit, les relations bidirectionnelles entre le muscle strié squelettique et les différents organes impactent le métabolisme à l’échelle du corps entier. Elles sont encore peu décrites dans le contexte de la DM1, mais représentent donc un aspect important pour la compréhension de ses mécanismes physiopathologiques. À l’échelle cellulaire, le métabolisme est défini comme l’ensemble des réactions biochimiques qui se déroulent dans la cellule pour en assurer l’homéostasie. Cinq grandes fonctions métaboliques sont classiquement définies : la production d’énergie, la biosynthèse de macromolécules, l’élimination de ces macromolécules et d’organites, la production et l’extraction de substrats, le renouvellement des composés cellulaires. Dans la DM1, les altérations de l’épissage dérégulent de nombreuses voies de signalisation impliquées dans le métabolisme cellulaire : les voies de la « protein kinase C » (PKC), du « nuclear factor-kappa B » (NF-kB), de la « glycogen synthase-kinase-3β » (GSK3β), du « calcineurin-nuclear factor of activated T-cells » (CnA-NFAT), de la « mammalian target of rapamycin » (mTOR), et celle de l’« adenosine monophosphate activated kinase » (AMPK) [7]. Plus précisément, la voie GSK3β exerce des effets pléiotropiques tels que le contrôle des mécanismes inflammatoires, oncogéniques, neurogéniques, myogéniques et métaboliques. Son hyperactivation dans la DM1 participe notamment aux défauts myogéniques des cellules souches musculaires (CSM) [8]. La voie CnA-NFAT est une voie clé de la plasticité musculaire, régulée par le signal calcique. L’hyperactivation de cette voie dans la DM1 se traduit par une transition vers une typologie plus oxydative du muscle. Ce changement de la typologie des fibres musculaires pourrait refléter un mécanisme compensatoire. La voie mTOR régule les niveaux d’anabolisme cellulaire, la synthèse protéique et les mécanismes autophagiques. Sa dérégulation favorise l’hyperactivation autophagique délétère pour le muscle DM1 [9]. Enfin, la voie de l’AMPK, principal senseur et régulateur du métabolisme cellulaire, est réprimée dans la DM1 [4, 9, 10]. Les conséquences potentielles de cette répression de l’AMPK sur le métabolisme cellulaire dans la DM1 sont discutées dans le paragraphe suivant.
Répression du régulateur du métabolisme cellulaire AMPK dans la DM1
L’AMPK est un senseur et un régulateur métaboliques, impliqué dans de nombreuses fonctions cellulaires telles que le cycle cellulaire, le catabolisme protéique et l’autophagie, le métabolisme du glucose, la biogénèse mitochondriale, la mitophagie et la dynamique mitochondriale. L’AMPK est une sérine/ thréonine kinase composée d’une sous-unité catalytique α et de deux sous-unités régulatrices β et γ, possédant de 2 à 3 isoformes chacune.
L’activation de l’AMPK dépend des niveaux énergétiques cellulaires, i.e. du ratio AMP/ATP, mais également de la phosphorylation d’AMPKα par la kinase LKB1 (liver kinase B1) ou par la kinase dépendante du calcium/ calmoduline kinase β (CaMKKβ). Lors d’un stress métabolique, la signalisation AMPK favorise les processus cataboliques pour régénérer l’ATP et inhibe les processus anaboliques consommateurs d’ATP [11]. Ainsi, l’AMPK fait le lien entre les régulations métaboliques et la myogenèse, et joue donc un rôle important dans la régénération musculaire : l’AMPKα1 dans la régulation métabolique du renouvellement des CSM [12] et l’AMPKα2 dans la régulation intrinsèque de l’accrétion myonucléaire (fusion myoblaste-fibre musculaire) [13].
Il s’avère que plusieurs études ont montré une répression marquée de la signalisation AMPK dans le muscle strié squelettique de souris DM1, dans un modèle de fibroblastes humains convertis en myoblastes [10], ainsi que dans des biopsies de muscle squelettique de patients [9]. Or, les mécanismes qui sous-tendent cette répression de l’AMPK dans la DM1 sont encore inconnus. Néanmoins, les activateurs pharmacologiques de l’AMPK ainsi que l’exercice physique ont induit un bénéfice non seulement sur l’histologie du muscle strié squelettique, mais également sur les anomalies d’épissage, dans plusieurs modèles de DM1 [4, 14]. La réactivation de la voie AMPK pourrait donc être bénéfique via ses multiples actions sur le métabolisme cellulaire et la production énergétique, par une amélioration des capacités myogéniques des CSM et donc de la régénération musculaire, mais aussi au travers d’un effet direct de l’AMPK sur le métabolisme des ARN. En effet, l’AMPK peut interagir et phosphoryler une protéine régulatrice de l’épissage alternatif, appelée ribonucléoprotéine nucléaire hétérogène H (hnRNP H). Cette protéine a la capacité de se lier aux CUGexp et participe donc à leur rétention nucléaire toxique [4]. L’activation de l’AMPK pourrait donc diminuer les foyers de CUGexp et moduler directement ce mécanisme physiopathologique à l’origine de la DM1.
Il reste encore de nombreuses zones d’ombre en ce qui concerne les mécanismes physiopathologiques de la DM1. Les conséquences des altérations de l’épissage sur le métabolisme cellulaire ne sont pas non plus caractérisées. Pourtant, plusieurs défauts métaboliques ont récemment été caractérisés dans la DM1, et toutes ces fonctions sont régulées par l’AMPK. Nous pouvons donc émettre l’hypothèse que la répression de l’AMPK joue un rôle dans ces dérégulations métaboliques et, plus encore, espérer une restauration de ces fonctions par les activateurs de l’AMPK.
Dysfonctions mitochondriales dans la DM1
La présence de mitochondries élargies et structurellement anormales avait déjà été décrite historiquement en microscopie électronique sur des biopsies musculaires de patients atteints de DM1, mais ces anomalies structurales mitochondriales n’ont été explorées que récemment. Parmi les centaines de transcrits anormalement épissés dans la DM1, une des signatures les plus altérées est celle du renouvellement et de la dynamique mitochondriaux [15, 16]. Un défaut d’expression des protéines mitochondriales ainsi qu’une altération de la balance fission/fusion ont également été décrits [16]. De plus, une diminution de la production d’ATP et de la phosphorylation oxydative, sans modification de la masse mitochondriale, a été décrite dans des fibroblastes de patients [17]. Enfin, un défaut d’épissage de l’OPA1 (optic atrophy type 1) qui code une protéine de fusion mitochondriale a été rapporté dans le muscle de souris DM1, et pourrait expliquer en partie ces altérations mitochondriales. L’AMPK régulant de multiples fonctions mitochondriales, sa répression dans la DM1 représente donc un mécanisme potentiel pouvant sous-tendre ces défauts métaboliques. À titre d’exemple, la metformine, un activateur indirect de l’AMPK et qui est un traitement approuvé du diabète de type II, a permis l’amélioration de la production d’ATP et de la respiration mitochondriale dans les fibroblastes de patients DM1 [17]. Autre exemple, l’exercice physique, qui est également un activateur de l’AMPK, a démontré un effet bénéfique sur la biogenèse, la dynamique et la respiration mitochondriales, dans des biopsies musculaires de patients [16]. Il existe donc aujourd’hui un faisceau d’arguments suggérant des altérations structurelles et fonctionnelles mitochondriales dans la DM1.
Un autre volet à explorer est l’entrée du calcium dans les mitochondries, qui est un processus important pour l’homéostasie cellulaire et pour la production mitochondriale d’ATP. Il est avéré que la signalisation calcique cytoplasmique est altérée dans la DM1, mais la signalisation calcique mitochondriale n’a jamais été étudiée. Pourtant, l’expression du canal calcique mitochondrial VDAC1 (voltage dependent anion channel 1) est diminuée dans la DM1, et améliorée par l’exercice [18], donc potentiellement via la réactivation de l’AMPK. L’entrée de calcium à la mitochondrie est non seulement régulée par VDAC1, mais également par son partenaire, le mitochondrial channel uniporter (MCU), dont la fonction est régulée par l’épissage alternatif et par l’AMPK [19]. L’altération simultanée de RYR et de VDAC, voire du MCU, dans la DM1 pourrait donc expliquer le déficit énergétique mitochondrial ; un déficit qui pourrait être modulé par les activateurs de l’AMPK. En outre, la signalisation calcique mitochondriale impacte également les processus autophagiques et mitophagiques, la dynamique mitochondriale ainsi que l’insulino-résistance. Il serait donc intéressant d’étudier le rôle de l’AMPK sur cette signalisation calcique mitochondriale et ses conséquences sur le métabolisme mitochondrial en général dans la DM1.
Dysfonctions du métabolisme du glucose dans la DM1
L’altération du métabolisme du glucose dans la DM1 est multifactorielle. Il existe tout d’abord un défaut d’absorption du glucose par la cellule musculaire du fait de l’altération de l’épissage du récepteur à l’insuline et du défaut de translocation à la membrane du transporteur du glucose GLUT4 [20]. Par ailleurs, il existe des anomalies d’épissage de la pyruvate kinase musculaire (PKM), une enzyme qui convertit le produit final de la glycolyse en pyruvate et en ATP, qui pourraient participer à l’altération du métabolisme du glucose et de la production d’ATP dans la DM1. Restaurer la voie AMPK dans la DM1 pourrait donc impacter l’insulino-résistance, l’absorption du glucose, la production d’ATP directement obtenue par la glycolyse, mais également la synthèse des macromolécules nécessaires aux régulations épigénétiques du métabolisme.
Altérations autoet mitophagiques dans la DM1
Dans la DM1, il existe également une forte activité autophagique qui participe à l’atrophie musculaire et aux défauts myogéniques [16]. Plusieurs acteurs clés des mécanismes autophagiques sont en effet dérégulés. La voie mTOR est anormalement activée. L’expression du gène FOXO-dépendant MYTHO (macroautophagy and youth optimizer) et de la sérine/thréonine kinase dépendante du calcium et de la calmoduline DAPK1 est diminuée. Enfin, cette altération de l’autophagie pourrait également être favorisée par les dysrégulations des signaux calciques. Or, l’exercice physique a permis de diminuer cette hyperactivation des mécanismes autophagiques chez des modèles murins. Cet effet bénéfique de l’exercice sur l’autophagie pourrait, une fois encore, être médié par la réactivation de l’AMPK.
Les processus mitophagiques sont, quant à eux, encore peu caractérisés dans la DM1. En revanche, plusieurs études ont montré une altération de la dynamique mitochondriale en faveur d’une augmentation de la fission [16]. L’altération de l’épissage d’OPA1, une protéine clé de la fusion mitochondriale, mais également de MFF, le facteur de fission mitochondriale, cible de l’AMPK, pourrait être impliquée dans l’altération du métabolisme mitochondrial et avoir de nombreuses conséquences sur la fonction musculaire et les capacités de régénération musculaire dans la DM1.
Une dérégulation de la mitophagie et de la dynamique mitochondriale peut favoriser la synthèse de niveaux toxiques d’espèces dérivées de l’oxygène (ROS) à l’origine du stress oxydatif et, en conséquence, altérer de nombreuses voies de signalisation. Un déséquilibre entre l’expression des gènes responsables d’un stress oxydatif et celle des gènes antioxydants a par ailleurs été mis en évidence à l’échelle transcriptomique dans la DM1. Restaurer les flux mitophagiques en activant l’AMPK pourrait donc permettre de lutter contre cet excès de stress oxydatif et prévenir ses mécanismes cytotoxiques.
« Booster » le métabolisme cellulaire dans la DM1, une approche innovante et prometteuse
Il est novateur de s’intéresser aux défauts métaboliques dans la DM1. Cette approche permet non seulement la caractérisation de la physiopathologie complexe de cette maladie, mais aussi l’identification de nouvelles stratégies thérapeutiques applicables à un large éventail de patients (Figure 1). Un arsenal thérapeutique pour activer la voie AMPK (activité physique, activateurs pharmacologiques, thérapies combinées) est déjà disponible en pratique clinique et de nouvelles molécules sont en cours de développement. Ces alternatives offrent de nouveaux espoirs à court terme pour les patients, avec des bénéfices attendus en termes d’épissage, de régénération musculaire, de production énergétique, et donc de fonction et de plasticité musculaire.
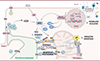 |
Figure 1 Pistes thérapeutiques liées aux altérations métaboliques dans la dystrophie myotonique de type I (DM1). Restaurer la voie de signalisation de l’AMPK dans les cellules musculaires DM1 pourrait permettre d’améliorer j la biogenèse mitochondriale, k la dynamique mitochondriale, l les processus mitophagiques et de régulation du stress oxydatif mitochondrial, m la signalisation calcique mitochondriale, n la production énergétique, o la sensibilité à l’insuline et le métabolisme du glucose. |
Prix SFM
Lola Lessard a reçu le prix du Meilleur poster lors des journées de la Société française de myologie (SFM) 2022.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Johnson NE, Butterfield RJ, Mayne K, et al. Population-Based Prevalence of Myotonic Dystrophy Type 1 Using Genetic Analysis of Statewide Blood Screening Program. Neurology 2021 ; 96 : e1045–e1053. [PubMed] [Google Scholar]
- Overend G, Légaré C, Mathieu J, et al. Allele length of the DMPK CTG repeat is a predictor of progressive myotonic dystrophy type 1 phenotypes. Hum Mol Genet 2019 ; 28 : 2245–2254. [CrossRef] [PubMed] [Google Scholar]
- De Antonio M, Dogan C, Hamroun D, et al. Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Rev Neurol (Paris) 2016 ; 172 : 572–580. [CrossRef] [PubMed] [Google Scholar]
- Ravel-Chapuis A, Duchesne E, Jasmin BJ. Pharmacological and exercise-induced activation of AMPK as emerging therapies for myotonic dystrophy type 1 patients. J Physiol 2022 ; 600 : 3249–3264. [CrossRef] [PubMed] [Google Scholar]
- Dahlqvist JR, Ørngreen MC, Witting N, Vissing J. Endocrine function over time in patients with myotonic dystrophy type 1. Eur J Neurol 2015 ; 22 : 116–122. [CrossRef] [PubMed] [Google Scholar]
- Severinsen MCK, Pedersen BK. Muscle–Organ Crosstalk: The Emerging Roles of Myokines. Endocr Rev 2020 ; 41 : 594–609. [CrossRef] [PubMed] [Google Scholar]
- Ozimski LL, Sabater-Arcis M, Bargiela A, Artero R. The hallmarks of myotonic dystrophy type 1 muscle dysfunction. Biol Rev Camb Philos Soc 2021 ; 96 : 716–730. [CrossRef] [PubMed] [Google Scholar]
- Grande V, Hathazi D, O’Connor E, et al. Dysregulation of GSK3β-target proteins in skin fibroblasts of Myotonic Dystrophy Type 1 (DM1) patients. J Neuromuscul Dis 2021 ; 8 : 603–619. [CrossRef] [PubMed] [Google Scholar]
- Brockhoff M, Rion N, Chojnowska K, et al. Targeting deregulated AMPK/mTORC1 pathways improves muscle function in myotonic dystrophy type I. J Clin Invest 2017 ; 127 : 549–563. [CrossRef] [PubMed] [Google Scholar]
- Ravel-Chapuis A, Al-Rewashdy A, Bélanger G, et al. Pharmacological and physiological activation of AMPK improves the spliceopathy in DM1 mouse muscles. Hum Mol Genet 2018 ; 27 : 3361–3376. [CrossRef] [PubMed] [Google Scholar]
- Steinberg GR, Hardie DG. New insights into activation and function of the AMPK. Nat Rev Mol Cell Biol 2023 ; 24 : 255–272. [CrossRef] [PubMed] [Google Scholar]
- Theret M, Gsaier L, Schaffer B, et al. AMPKα1-LDH pathway regulates muscle stem cell self-renewal by controlling metabolic homeostasis. EMBO J 2017 ; 36 : 1946–1962. [CrossRef] [PubMed] [Google Scholar]
- Kneppers A, Ben Larbi S, Theret M, et al. AMPKα2 is a skeletal muscle stem cell intrinsic regulator of myonuclear accretion. iScience 2023 ; 26 : 108343. [CrossRef] [PubMed] [Google Scholar]
- Misquitta NS, Ravel-Chapuis A, Jasmin BJ. Combinatorial treatment with exercise and AICAR potentiates the rescue of myotonic dystrophy type 1 mouse muscles in a sex-specific manner. Hum Mol Genet 2023 ; 32 : 551–566. [CrossRef] [PubMed] [Google Scholar]
- Todorow V, Hintze S, Kerr ARW, et al. Transcriptome analysis in a primary human muscle cell differentiation model for myotonic dystrophy type 1. Int J Mol Sci 2021 ; 22 : 8607. [CrossRef] [PubMed] [Google Scholar]
- Mikhail AI, Manta A, Ng SY, et al. A single dose of exercise stimulates skeletal muscle mitochondrial plasticity in myotonic dystrophy type 1. Acta Physiol (Oxf) 2023 ; 237 (4) : e13943. [CrossRef] [PubMed] [Google Scholar]
- García-Puga M, Saenz-Antoñanzas A, Fernández-Torrón R, et al. Myotonic Dystrophy type 1 cells display impaired metabolism and mitochondrial dysfunction that are reversed by metformin. Aging (Albany NY) 2020 ; 12 : 6260–6275. [CrossRef] [PubMed] [Google Scholar]
- Di Leo V, Lawless C, Roussel MP, et al. Resistance exercise training rescues mitochondrial dysfunction in skeletal muscle of patients with myotonic dystrophy type 1. J Neuromuscul Dis 2023 ; 10 : 1111–1126. [CrossRef] [PubMed] [Google Scholar]
- Zhao H, Li T, Wang K, et al. AMPK-mediated activation of MCU stimulates mitochondrial Ca2+ entry to promote mitotic progression. Nat Cell Biol 2019 ; 21 : 476–486. [CrossRef] [PubMed] [Google Scholar]
- Renna LV, Bosè F, Brigonzi E, et al. Aberrant insulin receptor expression is associated with insulin resistance and skeletal muscle atrophy in myotonic dystrophies. PLoS One 2019 ; 14 : e0214254. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1 Pistes thérapeutiques liées aux altérations métaboliques dans la dystrophie myotonique de type I (DM1). Restaurer la voie de signalisation de l’AMPK dans les cellules musculaires DM1 pourrait permettre d’améliorer j la biogenèse mitochondriale, k la dynamique mitochondriale, l les processus mitophagiques et de régulation du stress oxydatif mitochondrial, m la signalisation calcique mitochondriale, n la production énergétique, o la sensibilité à l’insuline et le métabolisme du glucose. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.