")
")
| Issue |
Med Sci (Paris)
Volume 40, Number 10, Octobre 2024
|
|
|---|---|---|
| Page(s) | 725 - 727 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2024124 | |
| Published online | 25 October 2024 | |
Prédiction de structures biomoléculaires complexes par AlphaFold 3
Prediction of complex biomolecular structures by AlphaFold 3
1
Laboratoire de biochimie théorique, UPR 9080 CNRS, Université Paris Cité Paris France
2
CNRS EMR 9002 Biologie structurale intégrative, Inserm U1167 – Facteurs de risques et déterminants moléculaires des maladies liées au vieillissement (RID-AGE), Univ. Lille, Centre hospitalo-universitaire de Lille, Institut Pasteur de Lille Lille France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
**
This email address is being protected from spambots. You need JavaScript enabled to view it.
L’année 2021 a été marquée par la formidable concrétisation de la prédiction des structures tridimensionnelles des protéines à partir de leurs séquences en acides aminés grâce à AlphaFold 2 [1] et RoseTTAFold [2], des systèmes d’intelligence artificielle (IA) fondés sur l’apprentissage profond par réseaux de neurones. L’IA a d’ailleurs marqué la toute récente édition 2024 des prix Nobel avec les prix Nobel de physique et de chimie décernés respectivement pour le développement de l’apprentissage profond et son application dans la prédiction des structures des protéines. Cette innovation est un considérable accélérateur de recherche et un remarquable générateur d’hypothèses. Elle transcende les méthodes traditionnelles comme la cristallographie aux rayons X et la cryo-microscopie électronique, en fournissant des modèles structuraux précis qui guident et complètent l’interprétation des données expérimentales [3]. Les prédictions se font à une échelle inédite, avec plus de 200 millions de modèles disponibles dans la base de données AlphaFold hébergée par l’Institut européen de bio-informatique (EBI) de l’EMBL (European molecular biology laboratory) [4]. Bien que très performante, l’IA d’AlphaFold 2 a de nombreuses limitations comme l’impossibilité de prendre en compte les modifications post-traductionnelles (PTM), les ions, les ligands, l’ADN et l’ARN [5]. La nouvelle version, AlphaFold 3, publiée en mai 2024 [6], ainsi que son alter ego RoseTTAFold All-Atom [7], comblent en partie les lacunes d’AlphaFold 2 et de RoseTTAFold.
Que se cache-t-il sous le capot d’AlphaFold 3 ?
L’une des principales innovations d’AlphaFold 3 est donc sa capacité à modéliser une large gamme de complexes biomoléculaires incluant quelques ligands, les acides nucléiques, quelques ions et certaines modifications post-traductionnelles (Figures 1 et 2). La structure d’entrée du modèle et la représentation des coordonnées ont été réorganisées pour tenir compte de ces changements. Dans AlphaFold 2, les acides aminés sont considérés en un bloc. Cette approche peut s’étendre aux acides nucléiques (ADN, ARN) et même aux modifications post-traductionnelles, mais pas audelà des biomolécules. AlphaFold 3, en revanche, modélise aussi des systèmes qui ne sont que des collections d’atomes individuels. Cela permet au modèle de traiter des molécules nouvelles, notamment pour la conception de médicaments.
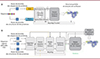 |
Figure 1. Architecture simplifiée de l’intelligence artificielle de AlphaFold 2 et AlphaFold 3. A. L’architecture d’AlphaFold 2 se décompose en trois modules principaux. Les données de séquences des protéines et les données expérimentales des structures 3D sont traitées par le module d’entrée, puis analysées par le module Evoformer, dont les résultats sont ensuite transmis au module de structure. B. L’architecture d’AlphaFold 3 se décompose en six modules principaux. Le module Pairformer d’AlphaFold 3 remplace le module Evoformer d’AlphaFold 2, et le module de diffusion remplace le module de structure. La représentation des alignements de séquences multiples (multiple sequence alignment, MSA) d’AlphaFold 2 (indiquée par la double flèche dans la figure 1A) n’a pas été conservée, et toutes les informations passent maintenant par la représentation matricielle par paires d’atomes ou d’acides aminés. Enfin, un module de confiance a été ajouté dans AlphaFold 3. Figure adaptée de [1, 6]. |
 |
Figure 2. Exemples de complexes biomoléculaires modélisés avec AlphaFold 3. Les modèles présentés ont été réalisés sur le serveur web AlphaFold 3 (https://alphafoldserver.com/), et les figures ont été réalisées avec ChimeraX. PTM : post-translational modification ; SP : résidu sérine phosphorylé. Les quelques ions et ligands disponibles sur le serveur web sont indiqués. |
Le module de structure d’AlphaFold 2 a été remplacé par un module de diffusion dans AlphaFold 3 (Figure 1). En tant que méthode générative, la diffusion modélise directement la génération des coordonnées atomiques, ce qui permet d’explorer des solutions très diverses. Pour éviter de générer des structures d’apparence plausible dans les régions non structurées, qui ne représentent pas fidèlement la réalité (ce qu’on appelle « hallucination »), la distillation croisée des modèles a été utilisée avec les données d’entraînement d’AlphaFold-Multimer [6]. Cette stratégie permet à AlphaFold 3 d’apprendre des succès et des échecs de son prédécesseur, et permet d’obtenir des prédictions plus précises. Le module Pairformer d’AlphaFold 3 remplace le module Evoformer d’AlphaFold 2 (Figure 1). L’importance accordée par AlphaFold 3 aux informations coévolutives extraites des alignements de séquences multiples (multiple sequence alignment, MSA) a été considérablement réduite. Enfin, un module de confiance a été introduit pour évaluer les erreurs provenant des calculs au niveau des atomes et des paires d’acides aminés (Figure 1). Certaines de ces modifications étaient nécessaires pour prédire avec précision les complexes biomoléculaires, tandis que d’autres sont des mises à jour des approches d’apprentissage automatique qui ont émergé et se sont répandues au cours des trois dernières années, depuis la publication d’AlphaFold 2.
Un nouveau champ des possibles
L’optimisation d’AlphaFold 2 élargit donc les applications d’AlphaFold 3 à presque toutes les molécules de la PDB (protein data bank) autres que les protéines (Figure 2). Par exemple, AlphaFold 3 surpasse les outils de docking protéine-ligand1 classiques tels qu’AutoDock Vina [8] (AlphaFold 3 atteint une performance de près de 80 %, tandis que celle d’AutoDock Vina est d’environ 55 %) et les outils de machine learning récents comme RoseTTAFold All-Atom sur le benchmark PoseBusters [9], qui contient 428 structures protéine-ligand de la PDB publiées avant 2021. AlphaFold 3 est également plus précis dans la prédiction des complexes protéineacide nucléique et des structures ARN que RoseTTAFoldNA [10] et AIchemy_RNA. En outre, AlphaFold 3 prédit plus précisément l’effet de ligands ou de certaines modifications post-traductionnelles des acides aminés et des bases d’acide nucléique (Figure 2). Cependant, aucune comparaison avec d’autres outils n’est rapportée. Le nombre de prédictions correctes, mesurées par un RMSD (root mean square deviation ; mesure utilisée pour comparer la qualité de la superposition entre la structure expérimentale et la structure prédite par AlphaFold 3) inférieur à 2 Å, varie entre 40 % pour les résidus modifiés de l’ARN et près de 80 % pour les ligands [6]. AlphaFold 3 améliore également la prédiction des complexes protéine-protéine par rapport à AlphaFold-Multimer, notamment celle des interfaces anticorps-protéine (performance de 63 % pour AlphaFold 3, contre 30 % pour AlphaFold-Multimer).
Des possibilités malheureusement limitées
Trois ans après sa sortie, l’IA d’AlphaFold 2 a été largement adoptée et maintes fois validée expérimentalement par la communauté scientifique comme un outil révolutionnaire de prédiction de structure. AlphaFold 3 est une belle optimisation d’AlphaFold 2, mais ce nouvel outil est limité à un usage scientifique non commercial. AlphaFold 3 obtient de très bons résultats dans toutes les catégories testées, mais il convient d’attendre les évaluations indépendantes des autres équipes scientifiques pour déterminer sa performance globale. Malheureusement, le code/modèle d’AlphaFold 3 n’est pas accessible comme l’était celui d’AlphaFold 2, l’accès étant limité à un serveur web qui impose des contraintes sur le nombre de modélisations par jour (20) et la taille des séquences d’entrée. Les modèles obtenus ne pourront pas être utilisés par la communauté académique pour faire du criblage virtuel par docking moléculaire ou entraîner d’autres modèles d’apprentissage automatique. Les autres restrictions concernent une liste très limitée de ligands (Figure 2). Compte tenu de toutes ces restrictions, il est très difficile, pour le moment, de déterminer les capacités de généralisation d’AlphaFold 3, en particulier pour les autres biomolécules qui ne sont pas contenues dans la liste prédéfinie.
Remerciements
Les auteurs souhaitent remercier Xavier Hanoulle ainsi que tous les membres de la section 20 du comité national de la recherche scientifique (CoNRS) pour les discussions fructueuses autour d’AlphaFold.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Ces outils de modélisation sont utilisés pour prédire en 3D la meilleure orientation d’un ligand (petite molécule) dans une protéine cible.
Références
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021 ; 596 : 583–9. [CrossRef] [PubMed] [Google Scholar]
- Baek M, DiMaio F, Anishchenko I, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021 ; 373 : 871–6. [CrossRef] [PubMed] [Google Scholar]
- Graille M, Sacquin-Mora S, Taly A. Best practices of using AI-based models in crystallography and their inpact in structural biology. J Chem Inf Model 2023 ; 63 : 3637–46. [CrossRef] [PubMed] [Google Scholar]
- Varadi M, Bertoni D, Magana P, et al. AlphaFold protein structure database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res 2024 ; 52 : D368–D375. [CrossRef] [PubMed] [Google Scholar]
- Versini R, Sritharan S, Aykac Fas B, et al. A perspective on the prospective use of AI in protein structure prediction. J Chem Inf Model 2024 ; 64 : 26–41. [CrossRef] [PubMed] [Google Scholar]
- Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024 ; 630 : 493–500. [CrossRef] [PubMed] [Google Scholar]
- Krishna R, Wang J, Ahern W, et al. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science 2024 ; 384 : eadl2528. [CrossRef] [PubMed] [Google Scholar]
- Eberhardt J, Santos-Martins D, Tillack AF, et al. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J Chem Inf Model 2021 ; 61 : 3891–8. [CrossRef] [PubMed] [Google Scholar]
- Buttenschoen M, Morris GM, Deane CM. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chem Sci 2024 ; 15 : 3130–9. [CrossRef] [PubMed] [Google Scholar]
- Baek M, McHugh R, Anishchenko I, et al. Accurate prediction of protein-nucleic acid complexes using RoseTTAFoldNA. Nat Methods 2024 ; 21 : 117–21. [CrossRef] [PubMed] [Google Scholar]
© 2024 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Liste des figures
|
Figure 1. Architecture simplifiée de l’intelligence artificielle de AlphaFold 2 et AlphaFold 3. A. L’architecture d’AlphaFold 2 se décompose en trois modules principaux. Les données de séquences des protéines et les données expérimentales des structures 3D sont traitées par le module d’entrée, puis analysées par le module Evoformer, dont les résultats sont ensuite transmis au module de structure. B. L’architecture d’AlphaFold 3 se décompose en six modules principaux. Le module Pairformer d’AlphaFold 3 remplace le module Evoformer d’AlphaFold 2, et le module de diffusion remplace le module de structure. La représentation des alignements de séquences multiples (multiple sequence alignment, MSA) d’AlphaFold 2 (indiquée par la double flèche dans la figure 1A) n’a pas été conservée, et toutes les informations passent maintenant par la représentation matricielle par paires d’atomes ou d’acides aminés. Enfin, un module de confiance a été ajouté dans AlphaFold 3. Figure adaptée de [1, 6]. |
| Dans le texte | |
|
Figure 2. Exemples de complexes biomoléculaires modélisés avec AlphaFold 3. Les modèles présentés ont été réalisés sur le serveur web AlphaFold 3 (https://alphafoldserver.com/), et les figures ont été réalisées avec ChimeraX. PTM : post-translational modification ; SP : résidu sérine phosphorylé. Les quelques ions et ligands disponibles sur le serveur web sont indiqués. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.