")
")
| Issue |
Med Sci (Paris)
Volume 39, Number 1, Janvier 2023
|
|
|---|---|---|
| Page(s) | 83 - 86 | |
| Section | Forum | |
| DOI | https://doi.org/10.1051/medsci/2022187 | |
| Published online | 24 January 2023 | |
Taille à l’âge adulte : une myriade de locus
Chroniques génomiques
Multiple loci account for human height
Biologiste, généticien et immunologiste, Président d’Aprogène (Association pour la promotion de la Génomique), 13007 Marseille, France
* This email address is being protected from spambots. You need JavaScript enabled to view it.
Abstract
The combination of 281 GWAS studies for adult height leads to the identification of 12,111 SNP in 7,209 loci that together account for essentially all SNP heritability for this trait: there is no longer any significant “missing heritability”, and these loci appear to be functionally relevant. This work is a model for future studies on the genetic determinants of complex diseases.
© 2023 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.

Après les premiers succès dans l’étude d’affections monogéniques, ou « mendéliennes », le séquençage du génome humain et l’invention de la technique des puces à ADN, ou microarrays, ont permis d’aborder l’analyse des déterminants génétiques de maladies multigéniques grâce aux balayages du génome ou GWAS (genome-wide association studies). Le travail publié en 2007 par le Wellcome Trust Case Control Consortium [1], sur sept affections fréquentes (de la maladie de Crohn au diabète en passant par l’arthrite rhumatoïde et la maladie bipolaire), portait au total sur 17 000 personnes étudiées grâce à des microarrays génotypant environ 500 000 SNP (single nucleotide polymorphism) – des effectifs considérables pour l’époque. Cette étude avait permis de retrouver les locus déjà identifiés pour ces affections et en révèla une bonne vingtaine de nouveaux, validant ainsi cette approche alors toute nouvelle. Dès lors, les GWAS allaient se développer, portant sur des effectifs croissants et révélant, à chaque fois, de nouveaux variants significatifs – mais avec une « rentabilité » décroissante, chacun d’eux expliquant une fraction de plus en plus faible de l’héritabilité. C’était logique, les variants les plus significatifs étant aussi les plus faciles à détecter, mais il semblait alors peu probable que l’accumulation de variants finisse par rendre compte de l’héritabilité des affections étudiées. Pour la maladie de Crohn, par exemple, les 71 locus identifiés au total en 2010 ne rendaient compte que d’environ 20 % de l’héritabilité de cette affection, telle que définie par l’étude des familles [2] (→).
(→) Voir la Chronique génomique de B. Jordan, m/s n° 3, mars 2011, page 323
On parlait alors d’« héritabilité perdue » (missing heritability) [3, 4] (→) et l’on échafaudait moult théories pour expliquer ce décalage [5]. L’analyse de caractères très multigéniques, mais relativement faciles à étudier, allait progressivement clarifier la situation.
(→) Voir les Chroniques génomiques de B. Jordan, m/s n° 5, mai 2010, page 541, et m/s n° 6-7, juin-juillet 2017, page 674
La taille, un cas d’école
La taille à l’âge adulte constitue un paramètre particulièrement intéressant de ce point de vue. Il s’agit d’un caractère facilement mesurable, répertorié dans toutes les bases de données, et, de plus, fortement héritable [6]. Dans les populations occidentales à l’abri de carences alimentaires, son héritabilité est estimée à 0,8 (ou 80 %) : grâce, notamment, aux études de vrais et faux jumeaux, on peut en effet affirmer qu’environ 80 % de la variance observée au sein de la population est due au patrimoine génétique des individus, le reste (20 %) étant lié à l’environnement. Et on sait depuis longtemps qu’il s’agit d’un caractère complexe impliquant de nombreux gènes. C’est donc un cas d’école pour des études GWAS approfondies, pour lesquelles il est clair que des effectifs importants seront nécessaires. Il faut noter ici que l’on ne peut pas s’attende à ce que les locus révélés par GWAS expliquent les 80 % d’héritabilité : les analyses génétiques (par microarray) sur lesquelles reposent ces études ne portent que sur le million de SNP examiné par les puces à ADN, et ces derniers ont été choisis pour leur polymorphisme ; on a éliminé les SNP très peu polymorphes (pour lesquels la fréquence de l’allèle mineur est inférieure à 1 %) qui, la plupart du temps, ne seraient pas informatifs1. On ignore donc les variants rares et leur contribution à l’héritabilité. Une étude pionnière parue en 2010 [7] suggérait que l’héritabilité de la taille liée à l’ensemble des SNP « communs » (non rares) était de l’ordre de 45 % ; cette estimation a été confirmée par la suite [8]. L’objectif de ces études est donc d’identifier un ensemble de SNP collectivement responsables d’une héritabilité d’environ 45 %. En 2018, une méta-analyse portant sur un total de 700 000 personnes [9] identifiait 3 290 SNP associés à la taille ; l’ensemble de ces SNP rendait compte d’environ 25 % de la variance de la taille, soit un peu plus de la moitié de l’héritabilité liée aux SNP. On a donc bien progressé, mais il restait encore la moitié du chemin à faire. C’est l’objet d’un article tout récent, qui rassemble cette fois des données portant sur près de cinq millions de personnes [10].
Des millions de personnes, et plus de douze mille SNP liés à la taille !
Ce travail, publié dans la revue Nature à l’automne 2022 [10], est l’œuvre d’un vaste consortium rassemblant plus de cinq cents laboratoires. Il s’agit en fait d’une méta-analyse regroupant 281 études GWAS qui portent au total sur 5,4 millions de personnes répertoriées soit dans le consortium GIANT (Genetic Investigation of ANthropometric Traits)2, soit dans la base de données de l’entreprise 23andMe, firme de « génomique récréative » qui a accumulé les millions de profils génétiques de ses clients [11] (→).
(→) Voir la Chronique génomique de B. Jordan, m/s n° 4, avril 2015, page 447
Comme le montre la Figure 1, cet échantillon est largement constitué d’individus d’origine européenne (plus de 75 %), un problème chronique dans les études GWAS qui commence tout juste à être pris en compte.
 |
Figure 1. Origine des populations étudiées dans le consortium GIANT (en %). EUR : European ; EAS : East Asian ; HIS : Hispanic ; AFR : African ; SAS : South Asian (extrait partiel de la figure 1 du supplément de [10]). |
Dans les différentes études GWAS intégrées dans la méta-analyse, le profil génétique de chaque ADN a été établi à l’aide de microarrays analysant environ un million de SNP – pas exactement les mêmes selon l’étude concernée et le type de microarray employé. Les auteurs de l’article ont déduit de la présente analyse le génotype pour l’ensemble des variants catalogués dans le projet HapMap 3 3, fournissant ainsi un balayage complet et cohérent des 5,4 millions de personnes étudiées. L’analyse des données pour les Européens a identifié près de dix mille SNP associés à la taille de manière significative ; l’étude des quatre autres populations a ajouté un peu plus de deux mille SNP supplémentaires, le total s’établissant à 12 111. Douze mille SNP pour la taille, cela fait beaucoup, et on pourrait se demander si, avec des analyses aussi poussées, on ne retombe pas sur le fait que chacun de nos gènes contribue, même un tout petit peu, à la taille à l’âge adulte. Il était donc nécessaire d’étudier comment ces SNP se répartissent sur le génome, combien de locus ils définissent, et quelle fraction du génome ils représentent.
Des SNP aux locus et aux gènes
Les auteurs ont donc défini une fenêtre de 100 kilobases (kb) centrée sur chacun des SNP identifiés, et ont examiné la présence d’un ou plusieurs autres SNP dans cette fenêtre. Il s’avère que 69 % des SNP sont à proximité d’un autre ; pour certains d’entre eux, le nombre de SNP co-localisés atteint la dizaine (avec un maximum de vingt-cinq). Avec ces regroupements, on arrive finalement à 7 209 locus couvrant environ 21 % du génome. Leur répartition est indiquée sur la Figure 2. On voit qu’elle est assez hétérogène avec des pics importants à certains endroits. On n’implique donc pas tous les gènes, mais un large sous-ensemble d’entre eux, a priori 7 000 environ, répartis sur l’ensemble des chromosomes.
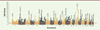 |
Figure 2. Densité des SNP (single nucleotide polymorphism) associés à la taille le long des chromosomes. Les locus présentant la densité la plus forte sur chaque chromosome ont été annotés avec le symbole du gène le plus proche. La moyenne et la médiane de la densité pour l’ensemble du génome sont figurées respectivement par les lignes rouges pleine et pointillée (extrait modifié de la figure 2 de [10]). |
Pour approcher la signification biologique de ces locus, les auteurs ont alors extrait de la base de données de génétique médicale OMIM (On-line Mendelian Inheritance in Man)4 462 gènes identifiés pour leur implication dans des anomalies de croissance osseuse, et ont constaté que les pics de densité de SNP étaient associés avec la présence de ces gènes, et cela d’autant plus que la densité de SNP était élevée. Le pic le plus important, situé sur le chromosome 15 et correspondant à 25 SNP regroupés dans une région de 700 kb, contient le gène ACAN (Aggrecan 1), codant le protéoglycane agrécane, un composant majeur de la matrice extracellulaire du cartilage. Ce gène est situé en 15q26.1 et les mutations qui l’affectent entraînent une dysplasie, une petite taille et un vieillissement du tissu osseux. Les associations repérées semblent donc bien biologiquement significatives.
L’héritabilité perdue… et retrouvée !
On peut alors calculer la fraction de l’héritabilité expliquée par cet ensemble de locus ; le calcul est fait pour chacune des populations étudiées, avec comme contrôle négatif, la fraction de l’héritabilité expliquée par l’ensemble des SNP situés en dehors des locus identifiés et représentant 79 % du génome. La Figure 3 montre les résultats : pour la population européenne (EUR), les 7 209 locus (soit 21 % du génome) rendent compte de la quasi-totalité de l’héritabilité liée aux SNP, avec un contrôle négatif pratiquement nul ; pour les quatre autres populations, les résultats, bien que significatifs, sont un peu moins nets, ce qui traduit le fait que leur faible effectif dans l’échantillon étudié (Figure 1) n’a pas permis le repérage de tous les SNP significatifs.
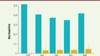 |
Figure 3. Héritabilité expliquée, pour chacune des cinq populations, par les 7 209 locus identifiés (21 % du génome, barres bleues), ou par le reste du génome (79 %, barres jaunes). En rouge : les barres d’erreur. EUR : European ; HIS : Hispanic ; SAS : South Asian ; EAS : East Asian ; AFR : African (extrait partiel et modifié de la figure 3 de [10]). |
Il n’y a donc plus d’héritabilité perdue, les 12 111 SNP (7 209 locus) identifiés rendent compte de l’ensemble de l’héritabilité liée aux SNP : comme l’indique le titre de l’article (A saturated map of common genetic variants associated with human height), on a réussi à saturer la carte, et la poursuite de ce type d’étude – du moins dans des populations européennes, déjà largement représentées – ne pourrait déceler de nouveaux variants significatifs. Reste à l’étendre à des échantillons plus importants d’autres populations, et, surtout, à creuser les implications fonctionnelles des locus découverts, comme dans le cas du gène ACAN cité ci-dessus.
Une étape importante
Les résultats rapportés ici montrent qu’il est possible d’identifier, par des analyses GWAS, l’ensemble des déterminants génétiques d’un caractère complexe – à condition d’y « mettre le prix », c’est-à-dire de faire porter l’étude sur un très grand nombre de participants. On voit aussi que le nombre de variants et de locus impliqués est très élevé, bien plus qu’on ne l’imaginait lors des premières publications. Il n’en reste pas moins que les localisations de ces variants sont bien corrélées avec celles des quelques centaines de gènes déjà connus pour influencer la taille : ils ouvrent donc autant de pistes pour identifier les processus biologiques impliqués.
Au-delà du remarquable exercice de style qu’il représente, cet article fournit aussi un modèle pour une étude plus approfondie d’affections multigéniques dont la part génétique est indéniable mais pour lesquelles les GWAS n’ont, jusqu’ici, donné que peu de résultats, avec une part considérable d’« héritabilité perdue ». Ce ne sera pas facile : les auteurs indiquent qu’il faudrait, par exemple, environ 35 millions d’individus pour arriver à la même saturation du génome pour la schizophrénie, un chiffre qui semble hors de portée. Reste aussi la question du rôle des variants rares, qui nécessitera le passage du microarray à la séquence intégrale, un important saut technique et surtout financier [12]. L’article de Yengo et al. [10] n’en constitue pas moins une étape importante et une belle démonstration de la puissance de l’approche GWAS, même dans des situations très complexes.
Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
On retomberait dans plus de 99 % des cas sur l’allèle majoritaire.
Qui a défini un « jeu minimum informatif » d’environ 500 000 SNP.Voir https://www.genome.gov/10001688/international-hapmap-project
Références
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007 ; 447 : 661–678. [CrossRef] [PubMed] [Google Scholar]
- Jordan B. Maladie de Crohn et GWAS, d’analyses en méta-analyses. Med Sci (Paris) 2011 ; 27 : 323–325. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Jordan B. À la recherche de l’héritabilité perdue. Med Sci (Paris) 2010 ; 26 : 541–543. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Jordan B. Variants fréquents et rares, caractères multigéniques et héritabilité perdue. Med Sci (Paris) 2017 ; 33 : 674–676. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature 2009 ; 461 : 747–753. [CrossRef] [PubMed] [Google Scholar]
- Galton F. Hereditary stature. Nature 1886: Jan 28: 295–8. https://www.york.ac.uk/depts/maths/histstat/galton_hereditary_stature.pdf. [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 2010 ; 42 : 565–569. [CrossRef] [PubMed] [Google Scholar]
- Wood A.R., Esko T., Yang J, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 2014 ; 46 : 1173–1186. [CrossRef] [PubMed] [Google Scholar]
- Yengo L, Sidorenko J, Kemper KE, et al. Meta-analysis of genome-wide association studies for height and body mass index in ~700000 individuals of European ancestry. Hum Mol Genet 2018 ; 27 : 3641–3649. [CrossRef] [PubMed] [Google Scholar]
- Yengo L, Vedantam S, Marouli E, et al. A saturated map of common genetic variants associated with human height. Nature 2022; 610 : 704–12. [CrossRef] [PubMed] [Google Scholar]
- Jordan B. 23andMe ou comment (très bien) valoriser ses clients. Med Sci (Paris) 2015 ; 31 : 447–449. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Wainschtein P, Jain D, Zheng Z, et al. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nat Genet 2022; 54 : 263–73. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Origine des populations étudiées dans le consortium GIANT (en %). EUR : European ; EAS : East Asian ; HIS : Hispanic ; AFR : African ; SAS : South Asian (extrait partiel de la figure 1 du supplément de [10]). |
| Dans le texte | |
|
Figure 2. Densité des SNP (single nucleotide polymorphism) associés à la taille le long des chromosomes. Les locus présentant la densité la plus forte sur chaque chromosome ont été annotés avec le symbole du gène le plus proche. La moyenne et la médiane de la densité pour l’ensemble du génome sont figurées respectivement par les lignes rouges pleine et pointillée (extrait modifié de la figure 2 de [10]). |
| Dans le texte | |
|
Figure 3. Héritabilité expliquée, pour chacune des cinq populations, par les 7 209 locus identifiés (21 % du génome, barres bleues), ou par le reste du génome (79 %, barres jaunes). En rouge : les barres d’erreur. EUR : European ; HIS : Hispanic ; SAS : South Asian ; EAS : East Asian ; AFR : African (extrait partiel et modifié de la figure 3 de [10]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.