")
")
| Issue |
Med Sci (Paris)
Volume 38, Decembre 2022
Les Cahiers de Myologie
|
|
|---|---|---|
| Page(s) | 52 - 54 | |
| Section | Fiche pratique | |
| DOI | https://doi.org/10.1051/medsci/2022184 | |
| Published online | 16 January 2023 | |
Dystrophie musculaire facio-scapulo-humérale
Vers un diagnostic moléculaire étendu à la FSHD2
Facio-scapulo-humeral muscular dystrophy: towards a molecular diagnosis extended to FSHD2
1
Aix Marseille Univ, INSERM Marseille Medical Genetics, Marseille, France
2
Département de Génétique Médicale, AP-HM, Hôpital d’enfants de la Timone, Marseille, France
3
Laboratoire Marseille Medical Genetics, U1251, INSERM ; Aix Marseille University. Faculté des Sciences Médicales et Paramédicales de la Timone. 27, Bd Jean Moulin 13005 Marseille, France
* This email address is being protected from spambots. You need JavaScript enabled to view it.
© Inserm
La dystrophie musculaire Facio-Scapulo-Humérale (FSHD) décrite par Landouzy et Déjerine en 1884 [1] est, en fréquence, la troisième myopathie en France après la dystrophie musculaire de Duchenne et la dystrophie myotonique de Steinert. Sa présentation clinique typique inclut une faiblesse progressive et asymétrique de certains muscles de la face, des ceintures scapulaire et pelvienne et des jambiers antérieurs. Son diagnostic est tardif, avec des symptômes apparaissant en moyenne entre 20 et 30 ans [2]. Dans environ 4 % des cas, elle peut se déclarer dans l’enfance, avec généralement des formes plus sévères [3,4].
Sa prévalence varie entre 1/ 8 000 et 1/ 12 000, et son incidence entre 0,38 et 0,7/ 100 000 habitants en fonction des populations de référence [5]. 3 000 personnes seraient ainsi concernées en France. La FSHD est une maladie à transmission autosomique dominante mais qui apparaît de novo dans 30 % des cas.
Dès le début des années 1990, des études de liaison ont associé la FSHD au locus 4q35 et plus précisément aux macrosatellites D4Z4. Chaque macrosatellite D4Z4 répété en tandem a une taille de 3,3 kilobases [6,9] (Figure 1). Dans la population générale, le nombre de D4Z4 au locus 4q35 varie entre 11 et 100 copies. La grande majorité des patients atteints de FSHD (95 %) portent entre 1 et 10 répétitions (dans la forme de FSHD de type 1 référencée FSHD1). Le locus 4q35 présente une forte homologie avec la région subtélomérique 10q26 qui comporte entre 1 et 100 répétitions D4Z4 mais dont le nombre n’est pas associé à la FSHD.
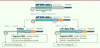 |
Figure 1. Schéma de la région 4q35 et processus associé au diagnostic moléculaire de la FSHD2. Représentation schématique du locus 4q35 : deux haplotypes existent dans la population. L’haplotype qB n’est pas associé à la FSHD. L’haplotype qA est caractérisé par la présence d’une région pLAM contenant un site de polyadénylation pour le transcrit DUX4 produit par la dernière répétition D4Z4. Dans la FSHD1, le nombre de répétitions est inférieur à 10. Les répétitions D4Z4 sont hypométhylées. Dans la FSHD2, le nombre de répétitions D4Z4 est supérieur à 10 unités mais celles-ci présentent une hypométhylation marquée. |
D4Z4 contient environ 70 % de dinucléotides GC. Dans la population générale, 65 à environ 80 % de ces dinucléotides sont méthylés et le niveau de méthylation est diminué dans la FSHD1 [10,11] (Figure 1).
Environ 5 % des patients (correspondant à la forme FSHD2 de la maladie) présentent un phénotype largement superposable à celui de FSHD1 mais sans réduction du nombre de répétitions D4Z4. En revanche, on y observe une hypométhylation marquée des loci 4q35 et 10q26. Les tableaux cliniques de FSHD1 et de FSHD2 ne diffèrent qu’en deux points : un âge de survenue plus tardif et une fréquence plus élevée de cas sporadiques (environ 60 %) pour la FSHD2 [12]. Des études d’exome entier (WES pour Whole Exome Sequencing) ont permis d’identifier, dans 85-90 % des cas de FSHD2 [13], des mutations du gène SMCHD1 (Structural Maintenance of Chromosomes flexible Hinge Domain-containing 1), codant une protéine de la famille des cohésines, et situé sur le bras court du chromosome 18.
Dans la FSHD1, le nombre résiduel de copies de D4Z4 est en grande partie inversement corrélé à la sévérité de la pathologie et à l’âge de survenue des symptômes. On note une apparition plus précoce et des signes plus marqués chez les patients porteurs de 1 à 3 unités. À l’inverse, on observe davantage de cas asymptomatiques, une survenue plus tardive ou des signes moins sévères pour les patients porteurs de 8 à 10 unités D4Z4. Le nombre de répétitions D4Z4 chez les patients FSHD2 serait réduit par rapport à la population générale et compris entre 11 et 20 unités [14]. Certains cas échappent toutefois à cette définition qui ne peut donc pas être appliquée de façon stricte. Celle-ci doit être réévaluée au cas par cas au regard du tableau clinique. La coexistence d’un nombre de répétitions D4Z4 aux alentours de 8-10 unités, intervalle qualifié de « zone grise », et d’une mutation de SMCHD1 a également été rapportée ce qui permettrait d’expliquer une plus grande sévérité ainsi qu’une variabilité clinique intrafamiliale plus importante[15].
La FSHD est également associée en cis à un haplotype noté 4qA, comportant un signal de polyadénylation (PAS) au niveau d’une région de 260 paires de bases (pLAM). Cet allèle, dit permissif, est présent dans 45 % de la population générale et diffère de son homologue 4qB, dit non permissif car ne contenant pas la séquence pLAM (Figure 1). Le pLAM serait nécessaire à la transcription de la dernière répétition de D4Z4 et la production d’un ARN messager (DUX4) polyadénylé. Le modèle physiopathologique actuel de la FSHD associe une réduction du nombre de répétitions D4Z4, ou des mutations de SMCHD1, à une hypométhylation de D4Z4, le tout entrainant l’activation d’une expression ectopique de DUX4 codé par la dernière répétition D4Z4 et la région pLAM, dont le produit, la protéine DUX4, serait toxique pour la cellule musculaire [16].
Une région polymorphe dans la partie proximale des loci 4q et 10q a également été décrite et certains polymorphismes (SSLP 159, 161, 168) associés à la FSHD [17]. Toutefois, sauf dans certains laboratoires, ceux-ci ne sont pas testés dans un but diagnostique et leur fonction exacte dans la maladie reste indéterminée.
Le Département de Génétique Médicale et Biologie Cellulaire (DGMBC) de la Timone est l’un des deux centres en France en charge du diagnostic moléculaire de la FSHD. Dans la majorité des cas, le diagnostic est réalisé par la technique dite du peignage moléculaire [18]. Celle-ci permet d’évaluer le nombre de répétitions D4Z4 sur chacun des deux allèles 4q et 10q ainsi que la présence des haplotypes distaux (qA ou qB). Cette approche permet de déterminer la réduction du nombre de répétitions D4Z4 sur le chromosome 4 et l’association à un haplotype de type A, définissant ainsi une FSHD1.
Jusqu’en 2020-2021, le diagnostic de la FSHD2 reposait sur l’association d’un phénotype clinique typique et l’absence d’allèle 4q raccourci. En 2021, le DGMBC a mis en place le diagnostic moléculaire de la FSHD2.
Grâce aux échanges du groupe de travail sur la FSHD coordonné au sein de la commission « recherche » de la filière de santé Maladies Rares neuromusculaires (Filnemus), nous avons élaboré un questionnaire accompagnant ces demandes de diagnostic. Ce questionnaire reprend quelques éléments de la description clinique des patients afin de préciser s’il s’agit ou non d’un diagnostic de certitude. Il recueille également des informations relatives à la ségrégation familiale et à un diagnostic moléculaire préalable de FSHD1. Dans le cas où celui-ci n’a pas été réalisé ou nécessite une meilleure résolution, il est réalisé par peignage moléculaire afin de déterminer la taille de la région des répétitions D4Z4 des chromosomes 4q et 10q, et d’identifier l’haplotype (qA/qB) correspondant. Le diagnostic moléculaire de la FSHD2 implique ensuite un séquençage de l’exome et une analyse des panels des gènes associés aux dystrophies des ceintures afin d’exclure une pathologie autre que la FSHD. Enfin, l’analyse comportera éventuellement la recherche de variants du gène SMCHD1, classifiés selon les recommandations de l’American College of Medical Genetics (ACMG) [19]. Cette recherche de variants dans SMCHD1 est précédée ou complétée par une analyse de la méthylation de l’ADN au sein du laboratoire de recherche Marseille Medical Genetics par séquençage de l’ADN modifié au bisulfite de sodium [20]. Cette étape, qui peut conditionner l’analyse mutationnelle, permet aussi de valider l’impact de chaque variant sur le niveau de méthylation de D4Z4. Il est à noter que le diagnostic de la FSHD2 n’est proposé ni en diagnostic prénatal ni en présymptomatique.
L’expérience acquise depuis plusieurs années, dans un cadre de projets de recherche puis dans un objectif de diagnostic moléculaire, nous a permis de déterminer que le niveau de méthylation de D4Z4 est inférieur à 40 % chez les patients porteurs d’un variant SMCHD1 [11]. Nous n’observons pas de corrélation entre le type de mutation, le nombre d’éléments répétés D4Z4 sur l’allèle 4q le plus court ou le niveau de méthylation, ni de seuil strict à 20 unités répétées pour l’allèle 4qA le plus court.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Tawil R, Van Der Maarel SM. Facioscapulohumeral muscular dystrophy. Muscle Nerve 2006 ; 34 : 1–15. [CrossRef] [PubMed] [Google Scholar]
- Goselink RJM, Schreuder THA, van Alfen N, et al. Facioscapulohumeral Dystrophy in Childhood: A Nationwide Natural History Study. Ann Neurol 2018 ; 84 : 627–637. [CrossRef] [PubMed] [Google Scholar]
- Dorobek M, van der Maarel SM, Lemmers RJ, et al. Early-onset facioscapulohumeral muscular dystrophy type 1 with some atypical features. J Child Neurol 2015 ; 30 : 580–587. [CrossRef] [PubMed] [Google Scholar]
- Klinge L, Eagle M, Haggerty ID, et al. Severe phenotype in infantile facioscapulohumeral muscular dystrophy. Neuromuscul Disord 2006 ; 16 : 553–558. [CrossRef] [PubMed] [Google Scholar]
- Deenen JC, Arnts H, van der Maarel SM, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 2014 ; 83 : 1056–1059. [CrossRef] [PubMed] [Google Scholar]
- van Deutekom JC, Wijmenga C, van Tienhoven EA, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet 1993 ; 2 : 2037–2042. [CrossRef] [PubMed] [Google Scholar]
- Wijmenga C, Sandkuijl LA, Moerer P, et al. Genetic linkage map of facioscapulohumeral muscular dystrophy and five polymorphic loci on chromosome 4q35-qter. Am J Hum Genet 1992 ; 51 : 411–415. [PubMed] [Google Scholar]
- Wijmenga C, Hewitt JE, Sandkuijl LA, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet 1992 ; 2 : 26–30. [CrossRef] [PubMed] [Google Scholar]
- Sarfarazi M, Wijmenga C, Upadhyaya M, et al. Regional mapping of facioscapulohumeral muscular dystrophy gene on 4q35: combined analysis of an international consortium. Am J Hum Genet 1992 ; 51 : 396–403. [PubMed] [Google Scholar]
- van Overveld PG, Lemmers RJ, Sandkuijl LA, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. 2003; 35 : 315–317. [Google Scholar]
- Roche S, Dion C, Broucqsault N, et al. Methylation hotspots evidenced by deep sequencing in patients with facioscapulohumeral dystrophy and mosaicism. Neurol Genet 2019 ; 5 : e372. [CrossRef] [PubMed] [Google Scholar]
- de Greef JC, Lemmers RJ, Camano P, et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology 2010 ; 75 : 1548–1554. [CrossRef] [PubMed] [Google Scholar]
- Lemmers RJ, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet 2012 ; 44 : 1370–1374. [CrossRef] [PubMed] [Google Scholar]
- Lemmers RJ, Goeman JJ, van der Vliet PJ, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet 2015 ; 24 : 659–669. [CrossRef] [PubMed] [Google Scholar]
- Sacconi S, Lemmers RJ, Balog J, et al. The FSHD2 Gene SMCHD1 Is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet 2013 ; 93 : 744–751. [CrossRef] [PubMed] [Google Scholar]
- Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010 ; 329 : 1650–1653. [CrossRef] [PubMed] [Google Scholar]
- Lemmers RJ, Wohlgemuth M, van der Gaag KJ, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet 2007 ; 81 : 884–894. [CrossRef] [PubMed] [Google Scholar]
- Nguyen K, Walrafen P, Bernard R, et al. Molecular combing reveals allelic combinations in facioscapulohumeral dystrophy. 2011 ; 70 : 4 627–633. [Google Scholar]
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015 ; 17 : 405–424. [CrossRef] [PubMed] [Google Scholar]
- Roche S, Dion C, Broucqsault N, et al. Methylation hotspots evidenced by deep sequencing in patients with facioscapulohumeral dystrophy and mosaicism. Neurol Genet 2019 ; 5 : e372. [CrossRef] [PubMed] [Google Scholar]
© 2022 médecine/sciences – Inserm
Liste des figures
|
Figure 1. Schéma de la région 4q35 et processus associé au diagnostic moléculaire de la FSHD2. Représentation schématique du locus 4q35 : deux haplotypes existent dans la population. L’haplotype qB n’est pas associé à la FSHD. L’haplotype qA est caractérisé par la présence d’une région pLAM contenant un site de polyadénylation pour le transcrit DUX4 produit par la dernière répétition D4Z4. Dans la FSHD1, le nombre de répétitions est inférieur à 10. Les répétitions D4Z4 sont hypométhylées. Dans la FSHD2, le nombre de répétitions D4Z4 est supérieur à 10 unités mais celles-ci présentent une hypométhylation marquée. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.