")
")
| Issue |
Med Sci (Paris)
Volume 38, Number 2, Février 2022
|
|
|---|---|---|
| Page(s) | 159 - 167 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2022010 | |
| Published online | 18 February 2022 | |
L’autophagie modulée par les virus
Un rôle dans la progression tumorale
Autophagy modulation by viruses: An important role in tumor progression
1
Inserm UMRS1138, Centre de recherche des Cordeliers, 15 rue de l’École de médecine, 75006 Paris, France
2
Sorbonne université, Univ Paris 6, France
3
Département de chirurgie thoracique, Hôpital Cochin, 24 rue du Faubourg Saint-Jacques, AP-HP, 75014 Paris, France
*
pierre-emmanuel.joubert@sorbonne-universite.fr
L’autophagie est un processus métabolique important pour maintenir l’homéostasie cellulaire à des moments critiques du développement et/ou en réponse à un stress environnemental. Cela est particulièrement pertinent dans le cas des cancers, pour lesquels il a été montré que l’autophagie a un impact important sur leur survenue et sur la croissance tumorale. D’une part, elle limite la transformation cancéreuse des cellules précancéreuses à un stade précoce, mais, d’autre part, elle favorise la survie et la prolifération cellulaires, les métastases et la résistance aux thérapies anti-tumorales dans les tumeurs plus avancées. L’autophagie peut être induite par une grande variété de stimulus extracellulaires et intracellulaires. Les infections virales ont souvent été associées à une modulation de l’autophagie, dont l’impact sur la réplication virale ou la survie des cellules infectées diffère selon le modèle étudié. Dans un contexte tumoral, certains mécanismes moléculaires complexes par lesquels la modulation de l’autophagie par les virus peut influencer le développement des cellules précancéreuses ou cancéreuses ont été révélés. Cette revue présente les découvertes récentes concernant les répercussions d’une perturbation de l’autophagie par l’infection virale sur la survenue et la progression des tumeurs cancéreuses.
Abstract
Autophagy is an important process for cellular homeostasis at critical steps of development or in response to environmental stress. In the context of cancers, autophagy has a significant impact on tumor occurrence and tumor cell growth. On the one hand, autophagy limits the transformation of precancerous cells into cancer cells at an early stage. However, on the other hand, it promotes cell survival, cell proliferation, metastasis and resistance to anti-tumor therapies in more advanced tumors. Autophagy can be induced by a variety of extracellular and intracellular stimulus. Viral infections have often been associated with a modulation of autophagy, with variable impacts on viral replication and on the survival of infected cells depending on the model studied. In a tumor context, the modulation of autophagy induced by the viral infection of tumor cells seems to have a significant impact on tumor progression. The aim of this review article is to present recent findings regarding the consequences of autophagy disturbance by viral infections on tumor behavior.
© 2022 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (http://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (http://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (© Laura Poillet-Perez).
L’autophagie : un mécanisme clé de l’homéostasie cellulaire
L’autophagie, un mot qui se réfère littéralement au fait de « se manger soi-même », définit un mécanisme catabolique de dégradation et de recyclage présent dans toutes les cellules eucaryotes. Ce processus peut être activé afin de cibler certaines molécules ou organelles défectueuses présentes dans le cytosol et de les délivrer aux lysosomes pour qu’ils les dégradent. Les composés issus de cette dégradation pourront être réutilisés par la cellule pour son fonctionnement. L’autophagie est donc un mécanisme crucial pour le maintien de l’homéostasie cellulaire. Elle permet aux cellules de survivre à des stress environnementaux, tels que le manque de nutriments ou l’hypoxie. Trois différents types d’autophagie existent selon la machinerie moléculaire mise en jeu : la macroautophagie, la microautophagie et l’autophagie par l’intermédiaire des chaperonnes [1]. La macroautophagie (que l’on appellera autophagie par la suite) est la plus étudiée ; son importance a été mise en évidence dans des contextes physiologiques mais aussi physiopathologiques. Elle comporte de multiples étapes, et débute par la formation d’une membrane, appelée phagophore (dont l’origine cellulaire peut varier selon les modèles étudiés), qui va peu à peu s’étendre pour former les autophagosomes, des vésicules à double membrane qui capturent des composants intracellulaires altérés (protéines ou organelles). Les autophagosomes fusionnent ensuite avec des endosomes et des lysosomes, ce qui entraîne la dégradation des éléments emprisonnés et le recyclage dans le cytosol des métabolites ainsi produits (→) (Figure 1).
(→) Voir aussi le numéro thématique Autophagie, m/s n° 3, mars 2017
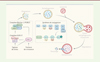 |
Figure 1. Le mécanisme de l’autophagie. Le complexe d’initiation des autophagosomes, formé de la bécline-1 et de VSP (vacuolar protein sorting) 34 (un membre de la famille de la phosphatidylinositol 3-kinase [PI3K]) joue un rôle crucial dans la formation et l’élongation du phagophore. L’activation de ce complexe, qui est inhibée ou stimulée par différents partenaires, produit, sur les membranes du phagophore, des groupements phosphatidylinositol 3-phosphate (PI(3)P1) permettant le recrutement de protéines nécessaires à l’apport de membranes au phagophore en croissance. L’élongation de ce dernier est assurée par le système de conjugaison constitué des protéines autophagiques (ATG) ATG12 et ATG5 à l’origine (avec ATG7 et ATG10) du complexe macromoléculaire ATG12-ATG5-ATG16 permettant la formation et le recrutement de LC3-II, formé de la conjugaison de LC3 (microtubule-associated protein 1A/1B-light chain 3) à la phosphatidyléthanolamine, responsable de l’élongation de l’autophagosome. La fermeture de la vésicule s’achève avec la libération des protéines ATG, et seul LC3-II lié à la membrane interne reste confiné à l’intérieur. La maturation de l’autophagosome est sous le contrôle de protéines facilitant les fusions vésiculaires, et la dégradation par les enzymes des lysosomes (figure créée avec BioRender.com). |
Bien que l’autophagie ait longtemps été considérée comme un processus de dégradation non sélectif, de nombreuses études ont montré qu’il pouvait être très spécifique grâce, notamment, à l’activité de protéines intermédiaires solubles permettant la liaison des protéines ou des organelles altérées au phagophore en formation (p62/SQSTM1 [sequestosome 1], NDP52 [nuclear dot protein of 52 kDa], OPTN [optineurin], NBR1 [next to BRCA1 gene 1]). C’est ainsi que la notion d’autophagie sélective a peu à peu émergé, et que sont apparues des désignations particulières selon les substrats ciblés : mitophagie, RE-phagie, ou encore virophagie, pour qualifier l’autophagie ciblant respectivement les mitochondries, le réticulum endoplasmique (RE), ou les composants viraux [1].
Depuis la mise en évidence de l’autophagie par le médecin biochimiste belge Christian de Duve en 19631, nos connaissances sur son mécanisme et sur son rôle physiologique ont beaucoup progressé. Elle est présente en permanence dans toutes les cellules eucaryotes, où elle agit comme un mécanisme de « contrôle qualité » en dégradant et en recyclant les protéines ou les organelles défectueux afin de maintenir l’homéostasie cellulaire. Il n’est donc pas très étonnant qu’un défaut d’autophagie soit associé, non seulement au vieillissement, mais aussi à diverses maladies humaines : maladies neurodégénératives, désordres métaboliques, susceptibilité aux infections, ou encore survenue et développement de cancers [2].
Autophagie et cancer
Rôle protecteur de l’autophagie contre la survenue de cancers
En raison de sa fonction homéostatique, de nombreuses recherches ont été réalisées pour déterminer l’impact de l’autophagie sur la survenue et le développement de cancers, mais cet impact reste difficile à évaluer dans ce contexte et semble très dépendant de l’environnement tumoral étudié. Plusieurs études s’accordent néanmoins pour définir l’autophagie comme un mécanisme suppresseur de tumeur dans les cellules saines, empêchant la survenue de cellules précancéreuses capables de devenir des tumeurs malignes [3]. En limitant la présence de mitochondries endommagées (ou d’autres organites défectueux), l’autophagie limite en effet l’apparition d’espèces réactives de l’oxygène pouvant causer des dommages à l’ADN cellulaire et entraîner la transformation des cellules saines en cellules cancéreuses. De nombreux patients souffrant de cancer présentent d’ailleurs des mutations de certains gènes impliqués dans l’autophagie. Le premier gène à avoir été considéré comme un gène suppresseur de tumeur est celui codant la bécline-1, dont la déplétion par mutation mono-allélique est observée dans de nombreuses tumeurs [4]. Depuis, l’altération de l’expression d’autres gènes (Bif-1 [Bax-interacting factor 1] ou UVRAG [UV radiation resistance- associated gene]) favorisant la mise en place de tumeurs a été observée dans les cellules tumorales. Des études réalisées in vitro ou dans des modèles murins ont également montré que l’inhibition du processus autophagique favorise la survenue de cancer, notamment de tumeurs solides, ce type de tumorigenèse étant associé à une production excessive d’espèces réactives de l’oxygène dans les cellules [5].
Rôle de l’autophagie sur la progression tumorale
Bien que l’autophagie puisse limiter la survenue de cancers, elle semble avoir un rôle opposé lorsque que la tumeur est établie, une forte activité autophagique des cellules cancéreuses pouvant être de mauvais pronostic. L’autophagie favorise en effet la survie des cellules tumorales en condition de stress, notamment dans les tumeurs solides dont les cellules sont soumises à l’hypoxie ou à un manque de nutriments, surtout lorsque la tumeur devient volumineuse, la néo-vascularisation étant alors plus faible au centre de la tumeur [6]. Carence nutritive et hypoxie induisent une forte réponse autophagique, ce qui permet la dégradation de composants cellulaires non essentiels afin de fournir les précurseurs métaboliques indispensables à la survie et à la prolifération des cellules cancéreuses. L’autophagie favorise également le développement de métastases en permettant à la cellule tumorale de résister au signal de mort cellulaire induit par sa perte d’adhérence à la matrice extracellulaire (anoikis), mais également en favorisant le maintien des cellules dans leur nouvelle niche, où se développera une tumeur secondaire [7].
L’autophagie a également un rôle important dans la résistance des cellules tumorales aux traitements anti-cancéreux conventionnels, tels que la chimiothérapie et la radiothérapie [8]. Certaines approches thérapeutiques consistent ainsi à combiner la chimiothérapie ou la radiothérapie et un inhibiteur de l’autophagie afin d’améliorer l’efficacité de ces traitements, avec des premiers résultats prometteurs [9, 10].
D’autres mécanismes expliquent le rôle protecteur de l’autophagie dans les tumeurs : le maintien des cellules souches cancéreuses, des cellules impliquées dans la résistance aux thérapies anti-cancéreuses, le développement de métastases, et la récurrence des cancers [11] ; l’échappement des cellules tumorales à la lyse par les cellules immunitaires (par les lymphocytes T cytotoxiques et les cellules NK [natural killer]), en induisant, dans les cellules cancéreuses, la dégradation des granzymes, des protéases que ces cellules de l’immunité produisent afin de les détruire [12].
Autophagie et virus
Une infection virale module l’autophagie
De nombreux facteurs peuvent stimuler le processus d’autophagie : la carence en nutriments ou en énergie, les stress du réticulum endoplasmique ou le stress oxydant, certaines cytokines, mais aussi l’infection par des microorganismes pathogènes. En effet, pour les virus par exemple, on sait aujourd’hui que de très nombreuses étapes de leur cycle réplicatif modulent l’activité autophagique de la cellule qu’ils infectent [13]. Leur simple reconnaissance par leur récepteur d’entrée peut être suffisante pour induire la formation d’autophagosomes. C’est notamment le cas pour le virus de la rougeole qui se lie à CD462 [14]. La reconnaissance des virus par les récepteurs de l’immunité innée peut également être à l’origine d’une stimulation de l’autophagie. Les récepteurs Toll-like (TLR), qui reconnaissent de nombreux composants protéiques ou nucléiques exprimés par les microorganismes pathogènes, ou les récepteurs RIG-I (retinoic acid-inducible gene-I)-like (RLR), qui reconnaissent l’ADN double brin cytosolique, sont ainsi à l’origine de la formation d’autophagosomes dans les cellules infectées [15]. Les virus peuvent également moduler la réponse autophagique des cellules qu’ils infectent par des mécanismes indirects. Les stress résultant de leur réplication (stress du réticulum endoplasmique ou stress oxydant) ou la production de cytokines pro-inflammatoires (interleukine[IL]-6, IL-1b, interféron[IFN]-g) ou anti-virales (IFN de type I), qui sont sécrétées en réponse à l’infection pour induire une réponse antivirale, sont autant de processus qui stimulent l’autophagie [16].
L’autophagie participe à la réponse anti-virale
L’autophagie, en recyclant les composants cytosoliques défectueux, peut également capturer et dégrader des constituants viraux présents dans le cytoplasme : particules ou protéines virales, mais également composants de la cellule nécessaires à la réplication des génomes viraux. Ce mécanisme de dégradation spécifique, appelé virophagie, attribue à l’autophagie un rôle important dans l’immunité antivirale, notamment dans les cellules non immunitaires. La première description de la virophagie a été réalisée dans le cadre de l’infection par le virus Sindbis3 : l’autophagie permet la dégradation d’une protéine de capside de ce virus, limitant ainsi sa propagation, donc la survenue de la maladie [17]. Ce mécanisme de dégradation spécifique a ensuite été décrit pour des infections par d’autres virus, notamment le virus de l’immunodéficience humaine de type 1 (VIH-1) [18].
L’autophagie peut également servir de relai pour la mise en place de réponses immunitaires antivirales efficaces, un rôle découvert dans le contexte de l’infection par le virus de la stomatite vésiculaire (VSV). L’autophagie favorise en effet le transport des antigènes viraux du cytosol vers des vésicules riches en TLR7, ce qui permet aux cellules infectées de reconnaître plus efficacement les virus et favorise la mise en place d’une réponse anti-virale adéquate [19]. L’autophagie participe également à la mise en place des réponses immunitaires adaptatives, notamment contre le virus Sindbis ou les Herpesvirus (dont le virus d’Epstein-Barr, EBV). Elle favorise la présentation des antigènes viraux en association avec les molécules du complexe majeur d’histocompatibilité de type II [20].
L’autophagie participe donc efficacement à la réponse antivirale. Néanmoins, la coévolution entre les virus et leurs cellules hôtes a permis à certains virus de développer des mécanismes permettant d’inhiber l’autophagie, voire de se servir des vésicules produites au cours du processus afin de favoriser leur réplication [21]. Certains virus empêchent le déclenchement de l’autophagie, ce qui leur permet de persister dans la cellule qu’ils ont infectée. De nombreuses protéines virales ciblent ainsi tout particulièrement la bécline-1, une protéine essentielle pour enclencher le mécanisme d’autophagie. C’est le cas notamment de HSV (Herpes simplex virus), KHSV (Kaposi’s sarcoma-associated herpesvirus), MHV (mouse hepatitis virus) ou HCMV (human cytomegalovirus). Certains virus (comme KHSV) peuvent également stimuler la voie de signalisation impliquant mTOR (mammalian target of rapamycin), un répresseur majeur de l’autophagie [22].
Une autre stratégie adoptée par les virus pour se protéger de la dégradation autophagique est le blocage de la fusion des autophagosomes avec les lysosomes. Cette stratégie a un double avantage pour les virus : non seulement elle les protège de la dégradation par les lysosomes, mais aussi elle leur fournit de très nombreuses vacuoles à double membrane, idéales pour leur réplication. Le premier virus pour lequel cette stratégie a été décrite est le virus de la poliomyélite [23]. Depuis, de nombreux virus se sont révélés capables de bloquer la phase de maturation de l’autophagie afin de bénéficier des vésicules à double membrane comme plateformes pour leur réplication.
Relations entre virus, autophagie et développement tumoral
Virus oncogéniques et autophagie
Bien que le cancer soit une maladie multifactorielle, combinant des prédispositions génétiques et des facteurs environnementaux, il est désormais établi qu’environ 15 % des cancers chez l’homme ont pour origine une infection virale, en particulier par les virus EBV (Epstein-Barr virus), HBV ou HCV (hepatitis B or C viruses), HPV (human papillomavirus), HTLV-1 (human T lymphotrophic virus of type 1), KHSV, ou MCPyV (Merkel cell polyomavirus) [24]. Bien que différents, aussi bien en termes de structure et de tropisme cellulaire que de cycle viral, ces virus sont tous à l’origine d’une infection chronique, un critère qui semble essentiel à leur pouvoir oncogénique.
Afin de se maintenir dans la cellule infectée, ces virus ont tous développé des mécanismes moléculaires pour stabiliser leur génome dans la cellule qui les héberge, empêcher sa mort, et échapper à la reconnaissance et à la lyse par les cellules immunitaires [24]. Des protéines impliquées dans le contrôle du cycle cellulaire, telles que p53 et pRb (protéine du rétinoblastome), sont parmi les cibles communes des oncoprotéines codées par le génome de ces virus. La dérégulation induite par ces virus apporte aux cellules infectées une capacité de proliféreration, alors incontrôlée, qui facilite le développement de la tumeur. L’intégration du génome viral dans celui de la cellule hôte modifie également l’expression de gènes qui codent des protéines participant à la régulation de l’homéostasie cellulaire, ce qui peut contribuer à la transformation d’une cellule normale en une cellule cancéreuse. Les virus oncogéniques ont ainsi développé des stratégies permettant l’inhibition de la phase d’initiation de l’autophagie, favorisant à la fois leur persistance dans la cellule hôte, mais également la tumorigenèse des cellules infectées. Le virus HPV de type 16, par exemple, inhibe l’autophagie de différentes manières : ses protéines, telles que HPV16 E5, E6 et E7, activent le répresseur de l’autophagie mTOR, inhibent le déclenchement de l’autophagie, et empêchent la fusion des autophagosomes avec les lysosomes [25]. Chez l’homme, on a ainsi montré que l’infection de cellules tumorales par HPV (notamment chez des patients atteints de cancer anal) diminuait leur activité autophagique ; dans des modèles murins, l’inhibition de l’autophagie induite par le virus augmente la survenue de tumeurs [26].
Les virus HBV et HCV, à l’origine du cancer du foie, ont également développé des mécanismes inhibant le processus d’autophagie, notamment la phase de dégradation, ce qui favorise leur réplication [27]. Chez des patients atteints d’un cancer hépatique, cette inhibition de l’autophagie dans les cellules tumorales infectées par HBV permet, entre autres, d’augmenter le niveau d’expression de miR-224, un micro-ARN qui favorise le développement du cancer. De même, la protéine non structurale NS4B du virus HCV inhibe la phase terminale du processus autophagique. Ceci conduit à l’accumulation de la protéine autophagique p62, ce qui stimule la voie dépendant du facteur de transcription Nrf2 (nuclear factor [erythroid-derived 2]-like 2) et favorise la tumorigenèse des cellules hépatiques infectées par le virus.
Le cycle des virus KHSV et EBV se décompose en deux phases : une phase de latence et une phase lytique. Leurs propriétés oncogéniques sont liées à la phase de latence, caractérisée par une diminution importante de l’activité autophagique de la cellule infectée. KHSV inhibe le processus en ciblant différentes molécules régulatrices et modulatrices de la cascade autophagique. La subversion de l’autophagie induite par le virus perturbe la sénescence cellulaire, ce qui favorise la prolifération des cellules infectées, deux conditions qui participent à la tumorigenèse cellulaire [28]. Néanmoins, le possible lien entre inhibition de l’autophagie et oncogenèse des cellules infectées reste à démontrer. Le virus EBV inhibe l’autophagie en activant le répresseur mTOR [29]. Cette perturbation est ainsi à l’origine d’une accumulation de p62 dans le noyau cellulaire, avec pour conséquences, un stress oxydant et une efficacité réduite de la réponse aux dommages de l’ADN, deux facteurs pouvant favoriser l’oncogenèse. Le virus HTLV-1, par l’action de sa protéine Tax-1, et le virus MCPyV réduisent, quant à eux, l’activité autophagique dans les cellules infectées en stimulant l’expression de plusieurs micro-ARN, favorisant l’apparition de cellules pré-cancéreuses [30].
Virus oncolytiques et autophagie
Certains virus sont à l’origine d’une régression tumorale. Ces virus, dits oncolytiques, ont été testés pour le traitement de certains cancers, notamment dans les cas de résistance acquise aux traitements conventionnels. Ces virus induisent une réponse anti-cancéreuse durable au cours du temps, car en plus d’infecter préférentiellement les cellules tumorales et de les lyser, ils provoquent la libération de molécules capables de stimuler la réponse immunitaire anti-tumorale [31]. Cette approche thérapeutique est particulièrement intéressante pour le traitement des tumeurs dites « froides », qui sont peu infiltrées par des cellules immunitaires [57] (→).
(→) Voir la Synthèse de J. Pol et al., m/s n° 2, février 2013, page 165
Virus oncolytiques et mort autophagique immunogène
Les virus oncolytiques exploitent différentes voies de mort cellulaire pour induire la lyse des cellules qu’ils infectent. Les parvovirus ou le virus NDV (Newcastle disease virus) induisent une apoptose dans de très nombreux cancers [32]. D’autres virus, comme les virus de la vaccine ou le virus de la variole, induisent préférentiellement une nécrose, notamment dans les cancers du côlon et de l’ovaire ou dans le mélanome [33]. Certains autres virus oncolytiques conduisent à une mort cellulaire qui dépend du processus autophagique.
Bien que l’autophagie ait été décrite comme un mécanisme par lequel la cellule compense les différents stress qu’elle subit afin de survivre dans des conditions défavorables, un excès d’autophagie peut lui être néfaste et conduire à sa mort. L’implication de l’autophagie dans la mort cellulaire programmée a été décrite pour la première fois dans une étude où des cellules ont été soumises à une carence nutritive importante et prolongée. Les auteurs ont remarqué la présence d’un nombre de vésicules autophagiques plus important dans les cellules mourantes que dans les cellules vivantes, suggérant qu’un stress prolongé serait responsable d’une hyperactivité autophagique associée à une consommation excessive de matériel cytosolique [34]. L’existence même d’un tel processus, appelé mort cellulaire dépendant de l’autophagie (dont l’abréviation en anglais est ADCD, pour autophagy-dependent cell death), fut longtemps contestée. L’impact fonctionnel de l’autophagie dans des cellules mourantes n’est en effet pas facilement démontrable : l’autophagie pourrait être aussi bien une réponse compensatoire de la cellule stressée, essayant de survivre, qu’un mécanisme qui agit directement, ou indirectement, sur l’induction d’une voie de mort cellulaire. Mais depuis ces premières descriptions, de très nombreuses études ont montré l’implication de l’autophagie dans l’induction de voies de mort cellulaire, soit directement en dégradant des inhibiteurs de ces voies (comme K-ras pour l’apoptose, la ferritine pour la ferroptose, la catalase pour la nécrose), soit indirectement, par l’utilisation des vacuoles autophagiques comme plateformes d’assemblage des complexes de mort cellulaire [34]. Toutes ces interrelations entre autophagie et voies de mort cellulaire ont contraint les chercheurs à préciser ce que recouvre le terme ADCD, aujourd’hui défini comme un type de mort cellulaire qui requiert l’action de composants moléculaires de la machinerie autophagique, et dont l’inhibition, génétique ou chimique, empêche la mort de la cellule [35].
Certains virus oncolytiques peuvent ainsi induire une ADCD. C’est notamment le cas des adénovirus, du virus NDV, du virus de la rougeole et du virus HSV dans de nombreux types de cancers [36]. Aujourd’hui, les thérapies fondées sur l’utilisation de virus oncolytiques visent à déclencher une ADCD afin de détruire plus efficacement les cellules tumorales. La stratégie la plus couramment utilisée pour stimuler l’ADCD consiste en l’ajout, dans le génome viral, d’un transgène qui permet la surexpression de la bécline-1 dans la cellule infectée. Cette stratégie a été appliquée au virus de la vaccine ou à des adénovirus, chez la souris, avec des résultats prometteurs dans les leucémies ou les myélomes, pour lesquels une diminution de la masse tumorale a été observée. L’implication de l’autophagie dans ce processus a été démontrée par sa disparition lorsqu’on inactive les gènes de l’autophagie [34].
Autophagie et mort cellulaire immunogène
Malgré leur capacité intrinsèque de lyse des cellules cancéreuses qu’ils infectent, les virus oncolytiques nécessitent l’induction d’une réponse immunitaire anti-tumorale pour avoir un réel impact dans le temps sur le développement des cancers. Cette capacité d’induire une réponse immunitaire à la suite de la lyse des cellules infectées est appelée mort cellulaire immunogène. Elle est caractérisée par l’exposition, à la surface des cellules mourantes, de la calréticuline (un signal « eat me », en français, « mange-moi ») et par la libération d’un cocktail de molécules, telles que des cytokines, des signaux de dangers (ATP, HMGB1 [high mobility group box 1], etc.), ou des antigènes. Tous ces éléments qui favorisent le recrutement et l’activation des cellules immunitaires dans le microenvironnement tumoral sont présents dans la majorité des cancers dont les cellules ont été lysées par des virus oncolytiques. Bien que les mécanismes impliqués dans la mise en place d’une réponse immunogène par les virus oncolytiques ne soient pas totalement élucidés, il semble que l’autophagie des cellules mourantes y contribue.
L’équipe dirigée par Beth Levine (1960-2020)4 a montré, dans un article princeps, que des cellules déficientes pour le mécanisme d’autophagie et mourantes ne sont pas reconnues et éliminées efficacement par les cellules immunitaires, contrairement aux cellules compétentes pour l’autophagie. Cet échappement immunitaire des cellules déficientes pour l’autophagie provient de leur incapacité d’exprimer à leur surface des signaux « eat me » de reconnaissance et de sécréter efficacement des molécules immunogènes [37]. Cette découverte sera ensuite confirmée dans différents modèles tumoraux, pour lesquels l’autophagie est requise pour la libération de molécules immunogènes après la lyse des cellules. L’équipe dirigée par Guido Kroemer a par exemple montré que seules les tumeurs compétentes pour l’autophagie sont capables d’attirer des cellules immunitaires et d’induire une réponse immunitaire anti-tumorale après traitement par différents composés cytolytiques [38]. Dans les cancers de la prostate, les adénovirus favorisent ainsi la libération d’ATP et d’HMGB1 par un mécanisme qui dépend de l’autophagie [39]. De même, dans les cancers du poumon, dans le glioblastome ou dans le mélanome, les virus NVD ou HSV induisent une mort immunogène via leur action sur l’autophagie des cellules qu’ils infectent, en augmentant notamment la sécrétion d’ATP, d’HMGB1 et d’HSP (heat shock protein) 79/90, mais aussi de certaines cytokines pro-inflammatoires : TNF-a (tumor necrosis factor a), IL-1b et GM-CSF (granulocyte macrophage colony-stimulating factor) [40]. Certains adénovirus sont également capables, en induisant une autophagie, de favoriser la présentation d’antigènes tumoraux via les molécules du complexe majeur d’histocompatibilité, augmentant ainsi l’activation de l’immunité anti-tumorale [41].
L’induction de l’autophagie dans des cellules cancéreuses infectées par des virus oncolytiques est donc impliquée dans la lyse directe des cellules tumorales, mais elle participe également à la mise en place d’une réponse immunitaire efficace limitant le développement tumoral. De nombreuses études sont actuellement en cours en vue d’une utilisation thérapeutique de virus oncolytiques recombinants capables d’induire plus efficacement le processus autophagique dans les cellules tumorales (en utilisant, par exemple, des adénovirus ou le virus de la vaccine exprimant la bécline-1) [42, 43].
Concordance de l’impact des infections virales et de l’autophagie sur la progression tumorale
Un nombre de plus en plus important d’études montrent l’impact des infections virales sur l’évolution des cancers. En plus des virus oncogéniques et oncolytiques, qui ont un rôle important dans la transformation tumorale et la lyse des cellules cancéreuses, d’autres virus peuvent participer à la progression tumorale, en influençant notamment la survie des cellules infectées, l’échappement immunitaire, l’angiogenèse, le développement de métastases ou la résistance aux traitements anti-cancéreux. Les mécanismes impliqués sont vraisemblablement divers. Néanmoins, de plus en plus de travaux révèlent l’existence d’une relation entre modification de l’autophagie par les virus et devenir des tumeurs.
Survie des tumeurs et modification de l’immunité anti-tumorale
De nombreux virus favorisent la survie des cellules qu’ils infectent en inhibant, notamment, l’apoptose ou d’autres voies de mort cellulaire programmée. Même si différents mécanismes peuvent être impliqués, l’autophagie semble jouer un rôle important. L’induction de l’autophagie dans les cellules infectées limite, par exemple, leur apoptose en réprimant l’expression des protéines pro-apoptotiques via la séquestration ou la dégradation des mitochondries, un processus notamment décrit dans le cas de cellules tumorales infectées. Dans des tumeurs pulmonaires, l’infection par le virus NDV induit en effet un mécanisme d’autophagie qui séquestre le cytochrome c (un facteur pro-apoptotique sécrété par les mitochondries) et empêche ainsi la mort des cellules tumorales [44]. Dans des cancers du cerveau, l’infection par le virus HTLV-1 induit une autophagie protectrice capable d’inhiber la mort cellulaire apoptotique induite par les récepteurs de mort, comme FAS-L (le ligand de la protéine Fas) et TRAIL (tumor-necrosis-factor-related apoptosis-inducing ligand) [45]. Plus récemment, il a été montré que l’induction d’une autophagie par le virus HBV favorisait la survie des cellules infectées et le développement du cancer du foie [46].
L’autophagie induite par une infection virale peut protéger les cellules cancéreuses non seulement de la mort cellulaire, mais également de l’effet de stress environnementaux ou d’attaques par le système immunitaire. En plus de limiter l’apoptose, la séquestration/dégradation sélective des mitochondries via l’autophagie permet de diminuer les réponses aux stress et la réponse immunitaire en limitant notamment la production d’espèces réactives de l’oxygène et la formation du complexe de l’inflammasome responsable de la production de cytokines pro-inflammatoires. L’autophagie induite par les virus peut également diminuer la production de cytokines en dégradant directement des facteurs cellulaires impliqués dans la réponse inflammatoire. L’infection par le virus HPV16 dans les cancers oro-pharyngés en fournit un exemple. En effet, dans les cellules tumorales infectées, l’autophagie induite par ce virus favorise la dégradation de STING (stimulator of interferon genes), un senseur cellulaire impliqué dans la production de cytokines inflammatoires en réponse aux virus [47]. La voie NF-kB (nuclear factor-kB) est également une cible de l’autophagie, avec pour conséquence, la perturbation de la réponse des cellules immunitaires du microenvironnement tumoral [48].
Les cellules cancéreuses sont également protégées de l’action des cellules immunitaires cytotoxiques par l’induction de l’autophagie, qui dans des conditions d’hypoxie, permet la capture et la dégradation des granzymes B produits par les lymphocytes T cytotoxiques et les cellules NK (natural killer) [49].
De même, dans les cancers du poumon, le déficit en protéines autophagiques réduit la phosphorylation de STAT3, un facteur de transcription important pour la résistance des tumeurs à la lyse par les lymphocytes T cytotoxiques [50]. Étant donné que de nombreux virus induisent l’activation de STAT3 dans les cellules infectées afin de favoriser la survie des tumeurs, il serait intéressant d’analyser le rôle de l’autophagie dans ce contexte.
Une autre stratégie favorisant l’échappement des cellules infectées par un virus à la lyse par les lymphocytes T cytotoxiques repose sur la réduction de l’expression des molécules du complexe majeur d’histocompatibilité de classe I (CMH-I) par les cellules infectées (par le virus VIH, le virus de la grippe ou le virus de la varicelle) ou par les cellules tumorales. Le rôle de l’autophagie dans ce processus n’est cependant pas clairement établi, bien qu’elle favorise l’endocytose des molécules du CMH-I et leur dégradation, et que son inhibition conduise à une présentation antigénique accrue, à la mise en place d’une réponse immunitaire plus importante et à une croissance tumorale fortement diminuée dans un modèle de cancer du pancréas [51].
Formation de métastases et résistance aux traitements anti-cancéreux
Les infections virales favorisent également l’angiogenèse, une étape majeure pour le développement des tumeurs, la dissémination des cellules tumorales dans l’organisme et le développement de métastases. Ce processus repose sur la production accrue de facteurs angiogéniques, comme le VEGF (vascular endothelial growth factor), et de facteurs chimio-attractants. Le virus HCV, en stimulant la production de CCL20 (C-C motif chemokine ligand 20), favorise la migration et l’invasion de cellules endothéliales dans la tumeur [52]. Les virus EBV, HPV, HSV2, HBV ou HCMV stimulent également l’angiogenèse et la formation de métastases de différents cancers. Comme l’infection virale, l’autophagie est un acteur important de la formation de métastases : elle favorise la survie des cellules tumorales lors de la transition épithélio-mésenchymateuse, ainsi que leur migration. Bien que le lien entre virus et induction autophagique dans le cadre du développement de métastases ne soit pas encore établi, une étude récente montre que, dans le cancer hépatique, l’infection par le virus HBV, via l’induction d’une autophagie, favorise l’expression du TGF-β (transforming growth factor β) et du lncRNA-ATB (long noncoding RNA activated by TGF-β), deux molécules impliquées dans la migration et l’invasion des cellules tumorales [53].
Les infections virales des cellules cancéreuses ont été associées à leur résistance à différents traitements anti-cancéreux, en inhibant l’apoptose induite par ces traitements, comme c’est le cas par exemple du virus EBV pour la chimiothérapie [54], ou des virus HPV16 et EBV pour la radiothérapie [55]. L’infection par le virus EBV, en activant la voie Akt (ou protéine kinase B), conduit également à une résistance des lymphocytes B au rituximab, un anticorps monoclonal dirigé contre ces cellules et qui induit leur lyse [56]. L’implication de l’autophagie dans les mécanismes de la résistance aux traitements anti-cancéreux associée aux infections virales reste cependant à déterminer. Notons néanmoins qu’autophagie et infection virale ont des effets similaires sur la résistance aux traitements, l’un modulant l’autre et tous deux inhibant principalement l’apoptose.
Conclusion
Selon le type et le stade de la maladie cancéreuse, le degré d’activité autophagique des cellules tumorales peut avoir différentes conséquences sur ces cellules. Au cours de l’étape initiale du développement tumoral, les cellules accumulent des dommages qui affectent les molécules impliquées dans le cycle cellulaire et favorisent le processus de tumorigenèse. Par ailleurs, les gènes suppresseurs de tumeurs et les oncogènes participent au contrôle de l’autophagie, reliant ainsi l’autophagie à l’apparition du cancer. Les virus oncogènes, qui conduisent à la transformation de cellules pré-cancéreuses en cellules cancéreuses, inhibent l’autophagie (Figure 2). Dans les cellules infectées, cette inhibition favorise l’accumulation d’organelles ou de protéines endommagées, à l’origine de molécules de stress pour la cellule et de mutations géniques. En revanche, plusieurs virus oncolytiques induisent un processus d’autophagie qui permet la reconnaissance des cellules tumorales mourantes par les cellules de l’immunité et leur élimination. À un stade plus avancé de la maladie, l’autophagie est susceptible de favoriser la croissance de la tumeur (Figure 2) en apportant aux cellules en train de proliférer les nutriments nécessaires. L’autophagie joue un rôle important dans la survie des cellules cancéreuses en favorisant notamment leur résistance à plusieurs types de thérapies. Elle peut également participer à la formation de métastases en protégeant de l’anoïkose, lors de leur migration dans les vaisseaux sanguins, les cellules tumorales qui se sont détachées de la tumeur primaire et qui iront établir de nouvelles colonies à distance. En modulant le processus autophagique, de nombreux virus vont ainsi agir sur le développement tumoral.
 |
Figure 2. Impact de la modulation de l’autophagie par les virus dans un contexte tumoral. Les infections virales activent (flèche verte) ou inhibent (flèche rouge) le processus d’autophagie dans les cellules tumorales. L’impact de ces modifications sur le développement de la tumeur est indiqué : les titres en vert correspondent à un effet anti-tumoral, et les titres en rouge à un effet pro-tumoral. LT : lymphocyte T ; NK : cellule natural killer ; EMT : epithelial-mesenchymal transition ; ROS : espèces réactives de l’oxygène (figure créée avec BioRender.com). |
La relation entre l’autophagie et le développement tumoral n’est pas établie. En revanche, l’existence d’un lien important entre l’autophagie et la perturbation du développement des tumeurs induite par les infections virales a été suggérée. Des études supplémentaires sont nécessaires pour comprendre les mécanismes reliant les infections virales et l’autophagie dans le contexte du développement tumoral. Cela pourrait déboucher, à terme, sur de nouvelles thérapies anticancéreuses et contre d’autres maladies marquées par une prolifération cellulaire.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- LevineB, KroemerG. Biological functions of autophagy genes: A disease perspective. Cell 2019 ; 176 : 11–42. [CrossRef] [PubMed] [Google Scholar]
- Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med 2020; 383 : 1564–76. [CrossRef] [PubMed] [Google Scholar]
- LiuEY, RyanKM. Autophagy and cancer: Issues we need to digest. J Cell Sc 2012 ; 125 : 2349–2358. [Google Scholar]
- LiangXH, JacksonS, SeamanM, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999 ; 402 : 672–676. [CrossRef] [PubMed] [Google Scholar]
- Liu W, Meng Y, Zong C, et al. Autophagy and tumorigenesis. Adv Exp Med Biol 2020; 1207 : 275–99. [CrossRef] [PubMed] [Google Scholar]
- Daskalaki I, Gkikas I, Tavernarakis N. Hypoxia and selective autophagy in cancer development and therapy. Front Cell Dev Biol 2018; 6. [PubMed] [Google Scholar]
- GuadamillasMC, CerezoA, del PozoMA. Overcoming anoikis: Pathways to anchorage-independent growth in cancer. J Cell Sci 2011 ; 124 : 3189–3197. [CrossRef] [PubMed] [Google Scholar]
- AmaravadiRK, KimmelmanAC, DebnathJ. Targeting autophagy in cancer: Recent advances and future directions. Cancer Discov 2019 ; 9 : 1167–1181. [CrossRef] [PubMed] [Google Scholar]
- ChenY, LiX, GuoL, et al. Combining radiation with autophagy inhibition enhances suppression of tumor growth and angiogenesis in esophageal cancer. Mol Med Rep 2015 ; 12 : 1645–1652. [CrossRef] [PubMed] [Google Scholar]
- ChengCY, LiuJC, WangJJ, et al. Autophagy inhibition increased the anti-tumor effect of cisplatin on drug-resistant esophageal cancer cells. J Biol Regul Homeost Agents 2017 ; 31 : 645–652. [PubMed] [Google Scholar]
- NazioF, BordiM, CianfanelliV, et al. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ 2019 ; 26 : 690–702. [CrossRef] [PubMed] [Google Scholar]
- ViryE, BaginskaJ, BerchemG, et al. Autophagic degradation of GZMB/granzyme B: a new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy 2014 ; 10 : 173–175. [CrossRef] [PubMed] [Google Scholar]
- ChoiY, BowmanJ, JungJ. Autophagy during viral infection: A double-edged sword. Nat Rev Microbiol 2018 ; 16 : 341–354. [CrossRef] [PubMed] [Google Scholar]
- JoubertP-E, MeiffrenG, GrégoireIP, et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009 ; 6 : 354–366. [CrossRef] [PubMed] [Google Scholar]
- KudchodkarSB, LevineB. Viruses and autophagy. Rev Med Virol 2009 ; 19 : 359–378. [CrossRef] [PubMed] [Google Scholar]
- JoubertP-E, WernekeS, de la CalleC, et al. Chikungunya-induced cell death is limited by ER and oxidative stress-induced autophagy. Autophagy 2012 ; 8 : 1261–1263. [CrossRef] [PubMed] [Google Scholar]
- OrvedahlA, MacPhersonS, SumpterR, et al. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010 ; 7 : 115–127. [CrossRef] [PubMed] [Google Scholar]
- SagnierS, DaussyCF, BorelS, et al. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J Virol 2015 ; 89 : 615–625. [CrossRef] [PubMed] [Google Scholar]
- LeeHK, LundJM, RamanathanB, et al. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007 ; 315 : 1398–1401. [CrossRef] [PubMed] [Google Scholar]
- PaludanC, SchmidD, LandthalerM, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005 ; 307 : 593–596. [CrossRef] [PubMed] [Google Scholar]
- JoubertP-E, AlbertML. Autophagy during viral infections: a double-edge sword. Virol Montrouge Fr 2013 ; 17 : 331–342. [Google Scholar]
- Bhatt AP, Damania B. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front Immunol 2012; 3. [PubMed] [Google Scholar]
- JacksonWT, Jr THG, Taylor MP, et al. Subversion of cellular autophagosomal machinery by RNA Viruses. PLOS Biol 2005 ; 3 : e156. [CrossRef] [PubMed] [Google Scholar]
- MesriEA, FeitelsonMA, MungerK. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014 ; 15 : 266–282. [CrossRef] [PubMed] [Google Scholar]
- BelleudiF, NanniM, RaffaS, et al. HPV16 E5 deregulates the autophagic process in human keratinocytes. Oncotarget 2015 ; 6 : 9370–9386. [CrossRef] [PubMed] [Google Scholar]
- CarchmanEH, MatkowskyjKA, MeskeL, et al. Dysregulation of autophagy contributes to anal carcinogenesis. PloS One 2016 ; 11 : e0164273. [CrossRef] [PubMed] [Google Scholar]
- KhanM, ImamH, SiddiquiA. Subversion of cellular autophagy during virus infection: Insights from hepatitis B and hepatitis C viruses. Liver Res 2018 ; 2 : 146–156. [CrossRef] [PubMed] [Google Scholar]
- LeidalAM, CyrDP, HillRJ, et al. Subversion of autophagy by Kaposi’s sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 2012 ; 11 : 167–180. [CrossRef] [PubMed] [Google Scholar]
- YinH, QuJ, PengQ, et al. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med Microbiol Immunol 2019 ; 208 : 573–583. [CrossRef] [PubMed] [Google Scholar]
- RenT, TakahashiY, LiuX, et al. HTLV-1 Tax deregulates autophagy by recruiting autophagic molecules into lipid raft microdomains. Oncogene 2015 ; 34 : 334–345. [CrossRef] [PubMed] [Google Scholar]
- Matos AL de, Franco LS, McFadden G. Oncolytic viruses and the immune system: The dynamic duo. Mol. Ther. - Methods Clin Dev 2020; 17 : 349–58. [CrossRef] [Google Scholar]
- KoksCA, GargAD, EhrhardtM, et al. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer 2015 ; 136 : E313–E325. [PubMed] [Google Scholar]
- WhildingLM, ArchibaldKM, KulbeH, et al. Vaccinia virus induces programmed necrosis in ovarian cancer cells. Mol Ther 2013 ; 21 : 2074–2086. [CrossRef] [PubMed] [Google Scholar]
- LinderB, Kögel D. Autophagy in cancer cell death. Biology 2019 ; 8 : 82. [Google Scholar]
- GalluzziL, VitaleI, AbramsJM, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ 2012 ; 19 : 107–120. [CrossRef] [PubMed] [Google Scholar]
- Jin K-T, Tao X-H, Fan Y-B, et al. Crosstalk between oncolytic viruses and autophagy in cancer therapy. Biomed Pharmacother 2021; 134 : 110932. [CrossRef] [PubMed] [Google Scholar]
- QuX, ZouZ, SunQ, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007 ; 128 : 931–946. [CrossRef] [PubMed] [Google Scholar]
- MichaudM, MartinsI, SukkurwalaAQ, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011 ; 334 : 1573–1577. [CrossRef] [PubMed] [Google Scholar]
- LiikanenI, AhtiainenL, HirvinenML, et al. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol Ther 2013 ; 21 : 1212–1223. [CrossRef] [PubMed] [Google Scholar]
- YeT, JiangK, WeiL, et al. Oncolytic Newcastle disease virus induces autophagy-dependent immunogenic cell death in lung cancer cells. Am J Cancer Res 2018 ; 8 : 1514–1527. [PubMed] [Google Scholar]
- KleinSR, JiangH, HossainMB, et al. Critical role of autophagy in the processing of adenovirus capsid-incorporated cancer-specific antigens. PloS One 2016 ; 11 : e0153814. [CrossRef] [PubMed] [Google Scholar]
- TongY, YouL, LiuH, et al. Potent antitumor activity of oncolytic adenovirus expressing beclin-1 via induction of autophagic cell death in leukemia. Oncotarget 2013 ; 4 : 860–874. [CrossRef] [PubMed] [Google Scholar]
- Lei W, Wang S, Xu N, et al. Enhancing therapeutic efficacy of oncolytic vaccinia virus armed with beclin-1, an autophagic gene in leukemia and myeloma. Biomed Pharmacother 2020; 125 : 110030. [CrossRef] [PubMed] [Google Scholar]
- MengG, XiaM, WangD, et al. Mitophagy promotes replication of oncolytic Newcastle disease virus by blocking intrinsic apoptosis in lung cancer cells. Oncotarget 2014 ; 5 : 6365–6374. [CrossRef] [PubMed] [Google Scholar]
- WangW, ZhouJ, ShiJ, et al. Human T-cell leukemia virus type 1 tax-deregulated autophagy pathway and c-FLIP expression contribute to resistance against death receptor-mediated apoptosis. J Virol 2014 ; 88 : 2786–2798. [CrossRef] [PubMed] [Google Scholar]
- MaoY, DaL, TangH, et al. Hepatitis B virus X protein reduces starvation-induced cell death through activation of autophagy and inhibition of mitochondrial apoptotic pathway. Biochem Biophys Res Commun 2011 ; 415 : 68–74. [CrossRef] [PubMed] [Google Scholar]
- Luo X, Donnelly CR, Gong W, et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J Clin Invest 2020; 130 : 1635–52. [CrossRef] [PubMed] [Google Scholar]
- FlissPM, JowersTP, BrinkmannMM, et al. Viral mediated redirection of NEMO/IKKγ to autophagosomes curtails the inflammatory cascade. PLOS Pathog 2012 ; 8 : e1002517. [CrossRef] [PubMed] [Google Scholar]
- ViryE, BaginskaJ, BerchemG, et al. Autophagic degradation of GZMB/granzyme B. Autophagy 2014 ; 10 : 173–175. [CrossRef] [PubMed] [Google Scholar]
- NomanMZ, JanjiB, KaminskaB, et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res 2011 ; 71 : 185976–5986. [CrossRef] [PubMed] [Google Scholar]
- Yamamoto K, Venida A, Yano J, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020; 581 : 100–5. [CrossRef] [PubMed] [Google Scholar]
- BenkheilM, Van HaeleM, RoskamsT, et al. CCL20, a direct-acting pro-angiogenic chemokine induced by hepatitis C virus (HCV): Potential role in HCV-related liver cancer. Exp Cell Res 2018 ; 372. [PubMed] [Google Scholar]
- Zhang Y, Li J, Wang S, et al. HBx-associated long non-coding RNA activated by TGF-β promotes cell invasion and migration by inducing autophagy in primary liver cancer. Int J Oncol 2020; 56 : 337–47. [PubMed] [Google Scholar]
- SeoJS, KimT-G, HongYS, et al. Contribution of Epstein-Barr virus infection to chemoresistance of gastric carcinoma cells to 5-fluorouracil. Arch Pharm Res 2011 ; 34 : 635–643. [CrossRef] [PubMed] [Google Scholar]
- WuQ, HanT, ShengX, et al. Downregulation of EB virus miR-BART4 inhibits proliferation and aggressiveness while promoting radiosensitivity of nasopharyngeal carcinoma. Biomed Pharmacother 2018 ; 108 : 741–751. [CrossRef] [PubMed] [Google Scholar]
- KimJH, KimWS, ParkC. Epstein-Barr virus latent membrane protein-1 protects B-cell lymphoma from rituximab-induced apoptosis through miR-155-mediated Akt activation and up-regulation of Mcl-1. Leuk Lymphoma 2012 ; 53 : 1586–1591. [CrossRef] [PubMed] [Google Scholar]
- PolJ, Le BœufF, DialloJS. Stratégies génétiques, immunologiques et pharmacologiques au service d’une nouvelle génération de virus anticancéreux. Med Sci (Paris) 2013 ; 29 : 165–173. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Le mécanisme de l’autophagie. Le complexe d’initiation des autophagosomes, formé de la bécline-1 et de VSP (vacuolar protein sorting) 34 (un membre de la famille de la phosphatidylinositol 3-kinase [PI3K]) joue un rôle crucial dans la formation et l’élongation du phagophore. L’activation de ce complexe, qui est inhibée ou stimulée par différents partenaires, produit, sur les membranes du phagophore, des groupements phosphatidylinositol 3-phosphate (PI(3)P1) permettant le recrutement de protéines nécessaires à l’apport de membranes au phagophore en croissance. L’élongation de ce dernier est assurée par le système de conjugaison constitué des protéines autophagiques (ATG) ATG12 et ATG5 à l’origine (avec ATG7 et ATG10) du complexe macromoléculaire ATG12-ATG5-ATG16 permettant la formation et le recrutement de LC3-II, formé de la conjugaison de LC3 (microtubule-associated protein 1A/1B-light chain 3) à la phosphatidyléthanolamine, responsable de l’élongation de l’autophagosome. La fermeture de la vésicule s’achève avec la libération des protéines ATG, et seul LC3-II lié à la membrane interne reste confiné à l’intérieur. La maturation de l’autophagosome est sous le contrôle de protéines facilitant les fusions vésiculaires, et la dégradation par les enzymes des lysosomes (figure créée avec BioRender.com). |
| Dans le texte | |
|
Figure 2. Impact de la modulation de l’autophagie par les virus dans un contexte tumoral. Les infections virales activent (flèche verte) ou inhibent (flèche rouge) le processus d’autophagie dans les cellules tumorales. L’impact de ces modifications sur le développement de la tumeur est indiqué : les titres en vert correspondent à un effet anti-tumoral, et les titres en rouge à un effet pro-tumoral. LT : lymphocyte T ; NK : cellule natural killer ; EMT : epithelial-mesenchymal transition ; ROS : espèces réactives de l’oxygène (figure créée avec BioRender.com). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.