")
")
| Issue |
Med Sci (Paris)
Volume 37, Decembre 2021
À Axel Kahn, pour ce qu’il fut
|
|
|---|---|---|
| Page(s) | 23 - 28 | |
| Section | Axel, l’homme de sciences et de communication | |
| DOI | https://doi.org/10.1051/medsci/2021222 | |
| Published online | 13 December 2021 | |
Axel Kahn à l’ère de la thérapie génique Les années 1980-2000
* This email address is being protected from spambots. You need JavaScript enabled to view it.

Axel Kahn (1944-2021).
À comparer les premiers articles de m/s sur la thérapie génique dans les années 1990 [1, 2] aux derniers articles sur ce thème [3–7], le vertige est patent : que de chemin parcouru, que d’écueils contournés et, au bout de la route, beaucoup de désillusions, mais aussi quelques magnifiques résultats ! L’enjeu était de taille, et les espoirs qu’elle suscitait étaient à l’avenant. Si les prémices de l’utilisation directe de l’ADN à des fins thérapeutiques dataient de 1970, dans les années 1990, cette approche relevait encore du magique, sorte de baguette qui permettrait d’effacer un défaut génétique, avec tout ce que ce diagnostic a d’implacable, pour y déposer, telle une marraine bienveillante au-dessus du couffin, un gène flambant neuf.
Il faut remettre ces premiers balbutiements dans le contexte de l’époque, si bien décrits dans un éditorial de Bertrand Jordan en 2006 [8]. L’ADN était alors devenu LE centre d’intérêt, les programmes Génome allaient bon train. Pourquoi alors, ne pas utiliser cette extraordinaire matière première, cette source infinie de protéines, pour laisser travailler la cellule et lui faire exprimer, à son corps consentant, l’enzyme d’intérêt, celle qui justement fait défaut lorsqu’une mutation génétique est venue en interrompre la séquence codante et, ainsi, réparer l’homme muté ? L’idée était simple, la réalisation fut plus complexe.
Revenons au contexte du laboratoire. En 1984, Axel Kahn prend la suite de Jean-Claude Dreyfus à la direction de l’unité de recherche de l’Inserm de génétique et de pathologie moléculaires. C’est l’ère de la génétique inverse, qui permet de découvrir de nouveaux gènes dont les mutations sont responsables de maladies humaines. On imagine qu’il en existe plus de 100 000 dans le génome humain et on coupe l’ADN en morceaux, on le clone, on l’exprime… Ces nouveaux outils donnent l’impression d’être soudain dotés de pouvoirs inouïs, c’est l’effervescence, c’est passionnant !

Les personnels de l’Institut de pathologie moléculaire réunis dans le cloître de la faculté de médecine Cochin-Port Royal en 1989.
Les premières souris dont des gènes ont été invalidés, qui vaudront par la suite le prix Nobel de physiologie ou médecine à Mario Capecchi et Oliver Smithies viennent de voir le jour. Une collaboration entre le laboratoire de Michel Perricaudet, pionnier français de la vectorisation adénovirale, et l ‘équipe de Pascale Briand, fraichement arrivée à Cochin, permet la correction à long terme d’un déficit en ornithine transcarbamylase chez la souris après administration d’un vecteur adénoviral portant une copie normale du gène [9]. En parallèle, le professeur Jean-Claude Kaplan, qui dirige le laboratoire de biochimie génétique et de diagnostic des maladies musculaires, a tissé des liens étroits avec l’AFM, l’Association française contre les myopathies et avec son président, Bernard Barataud. Le premier Téléthon avait démarré seulement quelques années auparavant, avec l’objectif de faire connaître les myopathies au grand public, de récolter des fonds pour aider les familles de malades et promouvoir la recherche de nouvelles pistes thérapeutiques. Avec l’équipe de Louis Kunkel aux Etats-Unis, découvreur du gène de la dystrophine, nous venons de mettre à jour les bases moléculaires permettant de distinguer dystrophie musculaire de Duchenne et dystrophie de Becker [10–12]. Dans la dystrophie de Duchenne, aucune protéine n’est synthétisée, car les mutations – le plus souvent des délétions – décalent le cadre de lecture, donnant lieu à un codon stop prématuré et à un transcrit instable. Dans la dystrophie de Becker, une protéine tronquée est synthétisée dans le muscle et la maladie s’en trouve atténuée. Ne disposant pas encore de nucléases ou de la technique CRISPR-Cas9 pour corriger en lieu et place la zone mutée, pourquoi ne pas essayer de réintroduire, à l’aide d’un vecteur adénoviral, une copie normale de l’ADNc muté chez les patients ? Avec une difficulté « de taille » cependant : le gène codant la dystrophine était le plus grand gène connu, record qui ne s’est pas démenti depuis, soit un gène de 2 300 kilobases, une protéine de 427 kilodaltons et un ARN messager de 14 kb, autant dire une gageure à faire entrer dans un vecteur adénoviral !
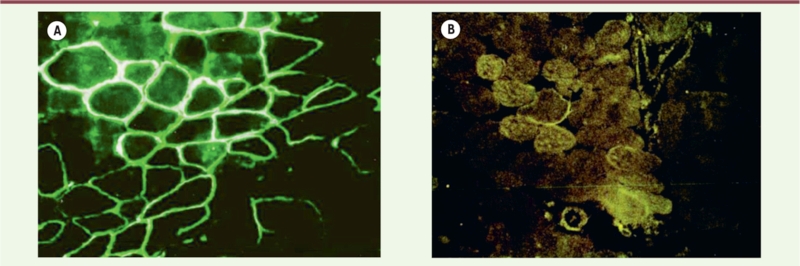
Correction du muscle de la souris dystrophique : à gauche, après injection d’un adénovirus exprimant une mini-dystrophine ; à droite, muscle non injecté. Coupe transversale du muscle injecté et immunohistochimie révélant la dystrophine à l’aide d’un anticorps fluorescent.
Il fallut l’énergie et l’enthousiasme communicatifs d’Axel Kahn pour fédérer ces trois équipes. Un étudiant en thèse du laboratoire de Michel Perricaudet, Thierry Ragot, une étudiante en thèse du laboratoire d’Axel, Nathalie Vincent, construisirent le vecteur adénoviral portant le gène d’une dystrophine tronquée, ou mini-dystrophine, de type « Becker » pour l’injecter dans le muscle de la souris mdx, modèle murin de la myopathie de Duchenne. Arrivée depuis peu dans le laboratoire de Kaplan, je fus associée à ce travail. Après quelques allers-retours entre Cochin et Villejuif, quelques mises au point avec des vecteurs traceurs, quelques longues séances d’injections sous hotte du vecteur viral contenant ou non la mini-dystrophine chez des souriceaux mutants, nous voilà prêts à analyser le résultat de ce transfert de gènes [13–15]. Je me souviens de cette fin d’après-midi où, sous le microscope à fluorescence, est apparu ce joli liseré vert qui marquait la dystrophine et était visible sur la quasi-totalité des fibres musculaires des muscles injectés, alors que celles des souris non traitées restaient d’un noir abyssal.

Axel Kahn dans son bureau, au sein de l’unité de recherche Inserm 129, au début des années 1990.
Axel Kahn était proche de ses étudiants, guettant les résultats, mémorisant parfaitement l’avancée des travaux. J’ai donc le souvenir de l’œil pétillant et ébloui d’Axel lorsqu’il posa les yeux sur le microscope. Et puisqu’il n’était pas avare d’encouragements, il a dû ponctuer ce résultat d’un classique « Haut les cœurs ! », aimant en toute circonstance user d’expressions qu’il répétait à l’envi et dont certaines comme « la plus belle fille du monde ne peut donner que ce qu’elle a », paraîtraient désormais « genrées » à défaut d’être misogynes !
Par la suite, d’autres corrections de phénocopies de maladies humaines chez la souris sont venues s’ajouter à cette « première », la sclérose latérale amyotrophique, dont l’atrophie musculaire progressive est ralentie par l’administration d’un vecteur viral par Thierry Bordet [16] ou par l’administration directe du gène exprimant la cardiotrophine par la dernière étudiante en thèse d’Axel, Jeanne Lesbordes [17], les premiers pas vers un traitement de l’hypercholestérolémie familiale par infection d’hépatocytes de souris et humains à l’aide d’un vecteur rétroviral, qui mèneront par la suite Anne Weber et son équipe à la modification des cellules fœtales hépatiques et, plus tard, à utiliser des cellules souches pluripotentes induites (iPS) humaines, long chemin semé d’embûches vers la thérapie cellulaire et génique de maladies génétiques hépatiques [18–21].

Axel Kahn et Alain Fischer lors du téléthon de l’an 2000.
Dès 1996, Axel Kahn restait cependant très prudent quant aux possibilités d’utiliser ces techniques chez l’homme et, dans l’enthousiasme de l’époque qui promettait grandeur et célébrité aux premiers essais cliniques qui s’avéreraient concluants, il mettait en garde : « les perspectives de traitement par thérapie génique de l’immense majorité des maladies génétiques restent extrêmement lointaines ». De fait, pour la myopathie de Duchenne, c’est il y a quelques mois seulement, en mars 2021, qu’une étude internationale portant sur une cinquantaine d’enfants de 6 à 10 ans a démarré, un premier garçon ayant reçu une injection d’un vecteur AAV (adeno-associated virus) de thérapie génique portant une micro-dystrophine à l’hôpital Trousseau.
Quels étaient les écueils, les contraintes identifiées par Axel [22] ? Le gène d’intérêt se devait d’être sous le contrôle de bonnes séquences régulatrices, capables de l’exprimer au bon niveau ; il fallait également disposer d’un vecteur et d’une voie d’administration qui permettraient d’introduire le gène thérapeutique dans les bonnes cellules et en quantités suffisantes, à moins que les cellules corrigées n’aient un avantage sélectif sur les cellules non corrigées. Tout cela, sans compter la réaction immunitaire induite par l’expression de la protéine codée par un gène et par l’introduction d’un vecteur viral inconnus du système immunitaire du patient. Bref, autant d’ingrédients d’une recette difficile à mettre en œuvre.
« Le grand homme en science, c’est d’abord celui qui sait discerner les bons problèmes au bon moment, quand il y a une chance de leur apporter une solution » écrivait François Jacob. Or, Axel était critique, lucide, mais également passionné et inventif. Il fit alors une proposition innovante fondée sur les résultats obtenus sur les souris SCID (severe combined immunodeficiency), qui donneraient lieu par la suite à la guérison par thérapie génique des premiers enfants immunodéprimés par l’équipe d’Alain Fischer et de Marina Cavazzana : donner un avantage sélectif aux cellules modifiées [23] en trouvant un organe capable de diffuser le gène thérapeutique efficacement.
Le foie paraissait un organe intéressant à cibler, étant non seulement facile d’accès mais pouvant également servir d’usine de production d’enzymes sécrétées dans la circulation. Cependant, contrairement au déficit immunitaire SCID où l’avantage sélectif est conféré directement par la copie normale du gène qui est muté chez les enfants immunodéficients, il fallait, en même temps que le gène d’intérêt, apporter l’avantage sélectif qui permettrait aux cellules modifiées de repeupler progressivement le foie, afin d’obtenir un effet thérapeutique quel que soit le déficit enzymatique et en dépit du faible nombre de cellules corrigées au départ. Il s’agissait donc de trouver un « couple infernal », formé d’un gène protecteur et d’un agent agressant le foie. Or, deux des étudiants en thèse d’Axel, Virginie Lacronique et Alexandre Mignon, venaient de publier dans la revue Nature Medicine que la surexpression du gène Bcl2 protégeait le foie d’une apoptose létale induite par l’activation de la voie Fas [24]. Axel suggéra alors d’utiliser ce couple Bcl2/Fas pour apporter la preuve de concept de l’efficacité de cette approche : l’idée était donc de transplanter le foie de souris normales avec des hépatocytes surexprimant Bcl2 (ou injecter directement in vivo un rétrovirus exprimant Bcl2) puis d’agresser de façon ménagée et répétée le foie des animaux ainsi traités par des doses sublétales de l’agent agoniste de la voie de Fas. Alors qu’elles représentaient moins de 1 % au départ, au bout de quelques semaines, les cellules initialement modifiées ou transplantées avaient repeuplé plus d’un tiers du foie [25–27] ! Mais il fallait aller plus loin que la preuve de principe, il fallait être thérapeutique. Axel nous suggéra alors de recueillir l’expertise d’Alain Tedgui, spécialiste des maladies vasculaires, qui disposait dans son laboratoire d’un modèle murin d’athérosclérose induite par le défaut en apolipoprotéine E, protéine majoritairement synthétisée par le foie, responsable d’une hypercholestérolémie. Ce fut une partie du travail de thèse de Claudia Mitchell de transplanter des hépatocytes normaux exprimant l’apolipoprotéine E et transduits par un vecteur viral pour leur faire exprimer également Bcl2, et de soumettre par la suite les animaux à une apoptose ménagée répétée du foie. Les quelques hépatocytes modifiés implantés au départ proliféraient alors sélectivement, aux dépens des cellules non corrigées. Et contrairement aux souris non traitées, celles-ci ne présentaient pas de plaques d’athérome [28, 29]. Le tour était joué et là encore, Axel sut partager notre satisfaction !

Repas post-séminaire dans la salle de conférence du dernier étage de la faculté de médecine Cochin.
Exigeant, parfois même intransigeant, lorsqu’on était un peu trop lent à suivre son cheminement de pensée, il n’était pas toujours tendre lorsque nous présentions nos résultats devant les membres de l’unité, et particulièrement devant lui, toujours assis au premier rang, qui ne cessait de nous interrompre en maniant l’imparfait du subjonctif avec délectation. Pendant ces séminaires, derrière l’écran de la grande salle de réunion du dernier étage de la faculté de médecine, se trouvait une toute petite cuisine, presque un couloir, où officiait Yvette, femme joviale et excellente cuisinière, si bien qu’Axel avait plaisir à dire qu’à défaut d’être les meilleurs de Paris, les séminaires de l’Institut étaient les plus odorants et les plus savoureux !

Axel Kahn décoré chevalier de la Légion d’honneur en 1995 par François Jacob.
Certes, étant issu de l’école des Schapira et Dreyfus, rompus à la génétique moléculaire, Axel nous encouragea à multiplier les modèles murins de maladies humaines, premier palier indispensable avant de proposer un essai clinique rigoureux, mais très vite, malgré les succès sur la souris, il déclara que ce type d’approche aurait probablement plus de débouchés dans des affections non génétiques. Or à ce jour, 65 % des indications visées par les essais cliniques déclarés de thérapie génique touchent des affections cancéreuses et, encore aujourd’hui, on peut se rendre compte que les espoirs suscités par cette approche pour les maladies génétiques sont à la hauteur des déceptions qu’elle engendre [30]. Le dernier article qu’Axel Kahn ait publié dans m/s sur la thérapie génique date de 2007, alors qu’il n’en était plus le rédacteur en chef depuis dix ans, juste avant qu’il ne quitte la direction de l’Institut Cochin pour la présidence de l’Université René Descartes. Il porte sur le traitement d’une maladie génétique effroyable, l’épidermolyse bulleuse [31]. Il avait probablement estimé le résultat suffisamment prometteur pour reprendre la plume et le style de m/s et en écrire une nouvelle… Curiosité encore, curiosité toujours…

Axel Kahn dans son bureau de la direction de l’Institut Cochin (2002-2007).
« Caractère à facettes variables, où se mêlaient une formidable mécanique intellectuelle (…), une absolue rigueur dans la critique, pour les autres comme pour lui-même ; un charme personnel qu’il modulait en fonction des situations, des interlocuteurs ; la faculté de se concentrer sur n’importe quel sujet et d’en changer dans la minute ; un certain goût de plaire, voire de se mettre en avant ; une totale disponibilité pour ses étudiants, une confiance en soi si manifeste qu’elle ne pouvait pas cacher une incertitude fondamentale, une immense gaîté avec un rire si communicatif, si retentissant qu’on pouvait immédiatement le localiser à distance ; un désir de dominer intellectuellement, voire de terroriser, qui le faisait s’installer au premier rang des séminaires pour bombarder de questions les conférenciers et leur expliquer, ou ce qu’ils avaient fait ou ce qu’ils auraient dû faire ; un enthousiasme à soutenir toute cause qu’il trouvait juste. Bref, une personnalité de feu (…) Opiniâtre et flamboyant ».
Si cela n’est pas le portrait d’Axel Kahn, puisqu’il s’agit du portrait que François Jacob fait de Monod dans « la Statue Intérieure », cela lui ressemble. En tous points.
Tout y est, la curiosité scientifique, l’intelligence, la rigueur critique, le goût pour la lumière, la passion, le rire si sonore, et peut être surtout, l’enthousiasme, essentiel pour mener un navire comme l’Institut Cochin, devenu un des plus gros centres de recherche publique française !

Jean-Claude Dreyfus et Axel Kahn, en pleine discussion scientifique à gauche, et lors d’un bal costumé de la première unité de recherches à droite.
Pourtant… « Une confiance en soi si manifeste qu’elle ne pouvait pas cacher une incertitude fondamentale… » Où se cachait cette incertitude fondamentale, alors qu’il était toujours sous contrôle jusqu’à ses coups de gueule qui étaient d’une fulgurante évidence, et ce d’autant plus que ses jugements rapides et incisifs en science étaient souvent exacts ? Je me souviens avoir vu ébranlée sa « Statue Extérieure » deux fois, au décès de Jean-Claude Dreyfus en 1995, ancien directeur de l’unité auquel il était très attaché, aussi réservé qu’Axel était solaire, mais d’une intelligence également remarquable, et dont Axel rappela l’histoire singulière dans médecine/sciences bien plus tard [32] ; puis, le 7 janvier 2015, alors qu’il avait déjà quitté ses fonctions à la présidence de l’Université Descartes, et était revenu travailler dans son bureau de l’Institut Cochin, jouxtant le mien, lorsqu’il en sortit brusquement, au bord des larmes, quelques minutes après les attentats de Charlie, en répétant « Ils ont tué Cabu ! Ils ont tué Cabu », sidéré, abasourdi.

De gauche à droite : Cabu, Axel Kahn, Michel Rencaud (président du festival « Rendez-vous du carnet de voyage »).
Agnostique, Axel disait : « Après la mort, il n’y a rien sauf peut-être le souvenir que vous pourrez garder de moi, et ça c’est déjà une forme d’immortalité »… à évoquer ces histoires anciennes, je sais combien les étudiants passés par ce laboratoire, dont le travail de seulement quelques-uns est relaté ici, ont été marqués par son amour de transmettre, par son énergie à défendre toute cause qu’il trouvait juste, et par cette manière si positive de faire émerger le meilleur d’eux-mêmes. La voilà cette part d’immortalité, la part des anges…

Hélène
Références
- Kahn A, Briand P. Nouvelles orientations pour la thérapie génique. Med Sci (Paris) 1990 ; 6 : 144–9. [CrossRef] [Google Scholar]
- Kahn A, Peschanski M. L’ADN, un médicament pour demain. Med Sci (Paris) 1992 ; 8 : 900–1. [CrossRef] [Google Scholar]
- Calvet C, Lahlou G, Safieddine S. Progrès de la thérapie génique. Espoirs pour le syndrome d’Usher. Med Sci (Paris) 2018 ; 34 : 842–8. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Jordan B. Hémophilie : la thérapie génique, enfin… Med Sci (Paris) 2018 ; 34 : 267. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Braun S. Thérapies géniques de l’amyotrophie spinale infantile – Un morceau d’histoire de la médecine. Med Sci (Paris) 2020 ; 36 : 141–6 [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Braun S. Biothérapies – Une révolution en marche. Med Sci (Paris) 2019 ; 35 (hors série n° 1) : 8–12. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Jordan B. Vers une thérapie génique pour la progéria. Med Sci (Paris) 2021 ; 37 : 413–6. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Jordan B. Thérapie génique grandes espérances. Med Sci (Paris) 2006 ; 22 : 451–2. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Stratford-Perricaudet LD, Levrero M, Chasse JF, et al. Evaluation of the transfer and expression in mice of an enzyme-encoding gene using a human adenovirus vector. Hum Gene Ther 1990 ; 1 : 241–56. [CrossRef] [PubMed] [Google Scholar]
- Koenig M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet 1989 ; 45 : 498–506. [PubMed] [Google Scholar]
- Gilgenkrantz H, Chelly J, Lambert M, et al. Analysis of molecular deletions with cDNA probes in patients with Duchenne and Becker muscular dystrophies. Genomics 1989 ; 5 : 574–80. [CrossRef] [PubMed] [Google Scholar]
- Gilgenkrantz H. Dystrophine, le charme discret d’une délétion. Med Sci (Paris) 1990 ; 6 : 308–9. [CrossRef] [Google Scholar]
- Ragot T, Vincent N, Gilgenkrantz H, et al. Transfert à l’aide d’un vecteur adénoviral d’un minigène de dystrophine dans des muscles de souris dystrophiques mdx. Med Sci (Paris), 1993 ; 9 : 238–41. [CrossRef] [Google Scholar]
- Ragot T, Vincent N, Chafey P, et al. Efficient adenovirus-mediated transfer of a human minidystrophin gene to skeletal muscle of mdx mice. Nature 1993 ; 361 : 647–50. [CrossRef] [PubMed] [Google Scholar]
- Vincent N, Ragot T, Gilgenkrantz H, et al. Long-term correction of mouse dystrophic degeneration by adenovirus-mediated transfer of a minidystrophin gene. Nat Genet 1993 ; 5 : 130–4. [CrossRef] [PubMed] [Google Scholar]
- Bordet T, Lesbordes JC, Rouhani S, et al. Protective effects of cardiotrophin-1 adenoviral gene transfer on neuromuscular degeneration in transgenic ALS mice. Hum Mol Genet 2001 ; 10 : 1925–33. [CrossRef] [PubMed] [Google Scholar]
- Lesbordes JC, Bordet T, Haase G, et al. In vivo electrotransfer of the cardiotrophin-1 gene into skeletal muscle slows down progression of motor neuron degeneration in pmn mice. Hum Mol Genet 2002 ; 11 : 1615–25. [CrossRef] [PubMed] [Google Scholar]
- Nguyen TH, Mainot S, Lainas P, et al. Ex vivo liver-directed gene therapy for the treatment of metabolic diseases: advances in hepatocyte transplantation and retroviral vectors. Curr Gene Ther 2009 ; 9 : 136–49. [CrossRef] [PubMed] [Google Scholar]
- Rashid ST, Corbineau S, Hannan N, et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest 2010 ; 120 : 3127–36. [CrossRef] [PubMed] [Google Scholar]
- Weber-Benarous A, Allain JE. Le foie fœtal : source incontournable de cellules souches hépatiques pour la thérapie cellulaire des maladies hépatiques ? Med Sci (Paris) 2002 ; 18 : 928–30. [CrossRef] [EDP Sciences] [Google Scholar]
- Touboul T, Vallier L, Weber A. Cellules souches embryonnaires humaines et iPS : une source fiable d’hépatocytes fœtaux. Med Sci (Paris), 2010, 26 : 1061–6. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Kahn A. Thérapie génique : le temps d’un premier bilan, Med Sci (Paris) 1996 ; 12 : 9–12. [Google Scholar]
- Fischer A, Hacein-Bey S, Le deist F, et al. Traitement du déficit immunitaire combiné sévère lié à l’X par transfert ex vivo du gène gamma c. Med Sci (Paris) 2000 ; 16 : 681–4. [CrossRef] [Google Scholar]
- Lacronique V, Mignon A, Fabre M, et al. Bcl-2 protects from lethal hepatic apoptosis induced by an anti-Fas antibody in mice. Nat Med 1996 ; 2 : 80–6. [CrossRef] [PubMed] [Google Scholar]
- Mignon A, Guidotti JE, Mitchell C, et al. Selective repopulation of normal mouse liver by Fas/CD95-resistant hepatocytes. Nat Med 1998 ; 4 : 1185–8. [CrossRef] [PubMed] [Google Scholar]
- Mignon A. Repopulation sélective du foie par des hépatocytes résistants à l’apoptose. Med Sci (Paris) 1998 ; 14 : 1147–8. [CrossRef] [Google Scholar]
- Guidotti JE, Mallet VO, Parlier D, et al. Fas/CD95 pathway induces mouse liver regeneration and allows for highly efficient retrovirus-mediated gene transfer. Hepatology 2001 ; 33 : 10–5. [CrossRef] [PubMed] [Google Scholar]
- Mitchell C, Guidotti JE, Mignon A, et al. Efficacité thérapeutique de la repopulation du foie dans un modèle murin d’hypercholestérolémie. Med Sci (Paris) 2000 ; 16 : 1007–9. [CrossRef] [Google Scholar]
- Mitchell C, Mignon A, Guidotti JE, et al. Therapeutic liver repopulation in a mouse model of hypercholesterolemia. Hum Mol Genet 2000 ; 9 : 1597–602. [CrossRef] [PubMed] [Google Scholar]
- Kwon D. Failure of genetic therapies for Huntington’s devastates community. Nature 2021 ; 593 : 180. [CrossRef] [PubMed] [Google Scholar]
- Kahn A. Thérapie génique d’une épidermolyse bulleuse jonctionnelle. Med Sci (Paris) 2007 ; 23 : 109–10. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Kahn A. Jean-Claude Dreyfus, un interne juif pendant la Guerre. Med Sci (Paris) 2009 ; 25 : 540–2. [CrossRef] [EDP Sciences] [Google Scholar]
© 2021 médecine/sciences – Inserm
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.