")
")
| Issue |
Med Sci (Paris)
Volume 37, Number 10, Octobre 2021
|
|
|---|---|---|
| Page(s) | 933 - 935 | |
| Section | Forum | |
| DOI | https://doi.org/10.1051/medsci/2021140 | |
| Published online | 14 October 2021 | |
Édition de gènes in vivo et thérapie génique
Chroniques génomiques
In vivo gene editing for gene therapy
ADÉS (Anthropologie bio-culturelle, droit, éthique et santé) UMR CNRS 7268, Aix Marseille université, Établissement français du sang.CoReBio PACA, case 901, Parc scientifique de Luminy, 13288 Marseille Cedex 09, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Abstract
In vivo gene editing has been achieved in a phase I clinical trial and results in a strong reduction of the level of a pathogenic protein. While preliminary, these results open the way for many applications in gene therapy.
© 2021 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.

Des débuts très laborieux
Les premières perspectives de thérapie génique sont apparues au cours des années 1970 [1], et les tentatives de réalisation se sont multipliées au cours des deux décennies suivantes. L’engouement pour cette approche était considérable, renforcé en France par l’action de l’Association française contre les myopathies (AFM) et son Téléthon annuel. Comme je l’ai rapporté dans une chronique il y a quelques années [2] (→), la revue L’usine nouvelle, émanation des milieux patronaux français, prévoyait en 1994 un marché annuel de 50 milliards de dollars américains pour 2010. En réalité le marché à cette date de la thérapie génique s’est avéré quasi nul, aucun des essais menés n’ayant donné de résultats vraiment concluants tandis que l’un d’eux entraînait en 1999 le décès d’un jeune volontaire, Jeffe Gelsinger. Au cours des années suivantes, les laboratoires impliqués se sont attachés à mieux comprendre les mécanismes cellulaires mis en jeu lorsqu’on introduit un vecteur portant un gène thérapeutique, à améliorer ces vecteurs et leurs modes d’administration, à augmenter l’expression des gènes transférés, et ces patientes mises au point ont enfin abouti à quelque vrais succès, notamment pour le traitement de l’hémophilie [3] (→).
(→) Voir la Chronique génomique de B. Jordan, m/s n° 10, octobre 2013, page 923
(→) Voir la Chronique génomique de B. Jordan, m/s n° 3, mars 2018, page 267
CRISPR en thérapie génique : premiers résultats
Entre-temps était apparue, à partir de 2012, une nouvelle technique sur laquelle beaucoup d’espoirs ont été immédiatement fondés. Il s’agit bien sûr du système CRISPR-Cas9 [4] (→) aussi appelé « Édition du génome » ou, de façon plus imagée « Ciseaux moléculaires ». Contrairement à la thérapie génique « classique », où l’on tente d’ajouter au génome du patient un exemplaire fonctionnel du gène en cause, cette technique offre en effet la possibilité de réellement corriger une mutation pathogène, de rétablir la séquence normale du gène, ou, éventuellement, de l’inactiver de manière spécifique [5] (→).
(→) Voir la Nouvelle de H. Gilgenkrantz, m/s n° 12, décembre 2014, page 1066
(→) Voir la Chronique génomique de B. Jordan, m/s n° 11, novembre 2015, page 1035
Parmi les premiers résultats positifs, il faut citer le travail d’une équipe nord-américaine [6] qui a traité des patients atteints de bêta-thalassémie ou d’anémie falciforme, en inactivant, grâce au système CRISPR-Cas9, un gène qui normalement réprime, à l’âge adulte, la synthèse de l’hémoglobine fœtale. Du coup, le patient synthétise ce type d’hémoglobine, ce qui traite efficacement l’affection. Deux bémols cependant : l’essai a porté sur un tout petit nombre de patients, et surtout, la modification par CRISPR-Cas9 se fait ex vivo, sur des cellules manipulées au laboratoire. Une fois modifiées in vitro, celles-ci doivent être réintroduites après élimination des cellules hématopoïétiques du malade par chimiothérapie – une procédure lourde et relativement risquée.
Le passage au traitement in vivo
L’approche ex vivo est évidemment limitée aux cas où les cellules dont on veut modifier l’ADN peuvent être prélevées, traitées in vitro puis réintroduites chez le malade. C’est loin d’être le cas général pour les affections héréditaires… Un pas vers l’in vivo a été franchi avec un traitement par CRISPR-Cas9 pratiqué par injection dans la rétine d’une construction visant à traiter l’amaurose congénitale de Leber en supprimant dans le gène CEP290 (centrosomal protein, 290-Kd) une mutation responsable de cette affection. L’essai est encore en cours, mais il est très prometteur [7]. L’article qui fait l’objet de cette chronique [8], lui, rapporte un travail plus avancé et fournit nombre d’informations précises. Il décrit le traitement par thérapie génique CRISPR-Cas9 in vivo d’une affection rare (de fréquence d’environ 1/50 000) mais grave, l’amylose à transthyrétine héréditaire. Cette maladie est causée par une mutation dans le gène TTR (transthyretin) qui code une protéine plasmatique, la transthyrétine (TTR), produite dans le foie et impliquée dans le transport de la vitamine A. La mutation provoque la synthèse d’une version anormale de cette protéine qui forme des fibrilles amyloïdes et cause des problèmes cardiaques graves entraînant le décès au bout de quelques années. L’affection est autosomique dominante, la présence de TTR anormale provoquant l’amylose, même en présence d’un excès de TTR normale. Comme plus de 100 mutations pathogènes différentes ont été répertoriées dans le gène TTR, il n’est pas possible de « réparer » ce gène en rétablissant la séquence normale ; l’objectif de la thérapie génique va être d’inactiver le gène TTR mutant pour éliminer la formation de fibrilles amyloïdes. Ce faisant, on va aussi inactiver le gène TTR normal situé sur l’autre chromosome, et donc abolir la synthèse de cette protéine ; mais il est possible de remédier à son absence en administrant au patient de la vitamine A.
Une mise au point très sérieuse
Le travail, rapporté dans un article du New England Journal of Medicine (NEJM) sous un titre sobre mais explicite, « CRISPR-Cas9 In Vivo Gene Editing For Transthyretin Amyloidosis », implique plusieurs laboratoires britanniques et néo-zélandais, ainsi que deux entreprises, Intellia Therapeutics et Regeneron Pharmaceuticals, toutes deux nord-américaines. Notons qu’Intellia est une société fondée en 2014 par Jennifer Doudna, co-découvreuse du système CRISPR-Cas9 avec la française Emmanuelle Charpentier [9] (→) et qui a pour objectif de développer les applications cliniques de cette technique. Une partie très importante de la mise au point a été consacrée aux nanoparticules lipidiques chargées de transporter le système CRISPR-Cas9 jusqu’à sa cible (Figure 1).
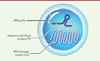 |
Figure 1. La nanoparticule lipidique utilisée pour l’édition de gènes in vivo (extrait partiel et modifié de la figure 1 de [8]). |
(→) Voir le Repères de B. Jordan, m/s n° 1, janvier 2021, page 77
Le produit à utiliser pour la thérapie génique, baptisé NTLA-2001, est donc constitué de nanoparticules qui renferment un ARN messager (de séquence adaptée aux cellules eucaryotes) codant l’enzyme Cas9, et un ARN guide contenant une séquence qui cible le gène TTR. Le tout est empaqueté dans une particule lipidique dont la composition précise est un secret industriel (proprietary) et qui assure, outre la protection de son contenu jusqu’à l’arrivée dans une cellule, un ciblage spécifique vers les hépatocytes qui sont le site quasi-exclusif de synthèse de la protéine TTR. Lors de l’entrée de la nanoparticule dans l’hépatocyte, l’enveloppe est dégradée, l’ARN messager Cas9 libéré est traduit pour donner la protéine Cas9 qui s’associe avec l’ARN guide. Le complexe ARN guide/Cas9 est alors internalisé dans le noyau et va effectuer une coupure double brin dans l’ADN au niveau du gène TTR (reconnu par l’ARN guide) ; les systèmes de réparation présents dans le noyau vont ensuite réparer cette coupure par jonction d’extrémités non homologues (NHEJ, non-homologous end joining), ce qui modifie le cadre de lecture de la protéine et, en général, abolit sa synthèse.
Tout ceci est bien beau en théorie, mais cela fonctionne-t-il réellement ? Les essais précliniques approfondis de NTLA-2001 donnent une réponse positive. Pratiqués dans un premier temps sur des hépatocytes humains en culture, ils ont montré l’inactivation du gène TTR et une réduction supérieure à 95 % du niveau de la protéine TTR ainsi qu’une absence de coupures parasites (off-target), même en présence de très fort excès du complexe ARN guide/Cas9. Des essais réalisés sur des souris transgéniques portant le gène TTR humain normal ou mutant ont confirmé que le système fonctionnait chez l’animal avec un effet perdurant au-delà de 12 mois. Point important : sur les souris traitées, une résection des 2/3 du foie suivie de sa régénération permet de voir que la quasi-abolition de synthèse de TTR se retrouve dans la partie régénérée, confirmant qu’il y a bien eu thérapie génique durable et non effet transitoire. Des études ultérieures réalisées sur le macaque (Macaca fascicularis) (avec un ARN guide adapté à la séquence du gène TTR de cette espèce) ont montré une réduction importante (supérieure à 94 %) et durable (au moins 12 mois) du taux de TTR sérique et ont permis d’évaluer la dose à utiliser pour les essais cliniques. Au total donc, un ensemble d’études précliniques sérieuses dont les résultats positifs permettent d’envisager le passage à un essai de phase I.
L’essai clinique : limité mais prometteur…
L’essai clinique a débuté fin 2020, avec des résultats prévus en 2022 et 2024 : les données de l’article commenté ici, publié début 2021, sont donc très préliminaires. Cet essai de phase I est destiné avant tout à évaluer la sécurité et les caractéristiques pharmacodynamiques du traitement. Il doit à terme inclure 38 patients, mais l’article ne concerne que les six premiers. Ces derniers ont reçu une seule infusion de NTLA-2001 à une dose de 0,1 mg/kg pour trois d’entre eux et de 0,3 mg/kg pour les trois autres. Leurs paramètres biologiques et la concentration de TTR dans leur sérum ont été suivis ensuite pendant au moins un mois. On a constaté très peu d’effets toxiques, moins fréquents à la dose la plus forte. En revanche, le niveau de TTR dans le sérum a fortement baissé (Figure 2), la baisse atteignant au bout de 28 jours environ 50 % du niveau initial pour les patients traités à la dose la plus faible, et 80 à 96 % pour ceux ayant reçu la dose la plus forte.
 |
Figure 2. Baisse progressive de la concentration sérique de la protéine TTR après infusion d’une seule dose de NTLA-1. Les doses employées (0,1 et 0,3 mg/kg) sont indiquées sur la figure ; les trois courbes de chaque panneau correspondent aux trois patients ayant reçu l’une ou l’autre dose de NTLA-1 (extrait partiel et modifié de la figure 4 de [8]). |
On a donc bien l’effet attendu : une baisse très significative de la concentration sérique de la protéine (normale ou mutante), observée sur plusieurs patients avec une cinétique de l’ordre du mois et dépendant de la quantité de produit NTLA-2001 administrée au malade. C’est donc une validation très nette de l’approche développée, obtenue dès le début d’un essai clinique de phase I. On comprend que les auteurs aient tenu à publier ces résultats malgré leur caractère préliminaire, car ils constituent une réelle validation de leur approche : l’édition de gènes in vivo semble effectivement fonctionner chez l’homme et aboutir au résultat biologique escompté.
Les suites
L’essai, prévu pour durer jusqu’en 2024 et devant inclure 38 patients, va naturellement se poursuivre. Reste à vérifier que le gène TTR a bien été inactivé chez les patients (c’est quasiment certain, mais il faut en être sûr), à augmenter le nombre de patients, et à vérifier que l’effet est durable. On peut déjà envisager les améliorations possibles du procédé : compte tenu de la bonne tolérance du traitement, on peut espérer voir une réduction plus forte encore du taux de TTR sérique en augmentant la dose de NTLA-2001. Mais surtout, il va falloir déterminer si cette thérapie aboutit, comme elle le devrait, à une amélioration clinique : arrêt de l’évolution négative des patients, peut-être même amélioration, si la TTR défectueuse peut s’éliminer progressivement ? Quoi qu’il en soit, ce premier aperçu d’un essai clinique à peine entamé montre que l’approche est prometteuse, que l’édition de gènes in vivo fonctionne, et il ouvre de réelles perspectives pour le développement des thérapies géniques – tout en stimulant l’intérêt des marchés financiers pour Intellia dont les actions ont doublé de valeur lors de la publication de cet article…
Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Friedmann TRoblin R. Gene therapy for human genetic disease?. Science 1972 ; 175 : 949–955. [PubMed] [Google Scholar]
- Jordan B. Un chapeau pour mon repas ?. Med Sci (Paris) 2013 ; 29 : 923–925. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Jordan B. Hémophilie : la thérapie génique, enfin. Med Sci (Paris) 2018 ; 34 : 267–270. [PubMed] [Google Scholar]
- La Gilgenkrantz H. révolution des CRISPR est en marche. Med Sci (Paris) 2014 ; 30 : 1066–1069. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Jordan B. CRISPR-Cas9, une nouvelle donne pour la thérapie génique. Med Sci (Paris) 2015 ; 31 : 1035–1038. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Frangoul H, Altshuler D, Cappellini D, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med 2021; 384 : 252–60. [PubMed] [Google Scholar]
- Ledford H. CRISPR treatment inserted directly into the body for first time. Nature 2020; 579 : 185. [PubMed] [Google Scholar]
- Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med 2021; 385 : 493–502. [PubMed] [Google Scholar]
- Jordan B. CRISPR : le Nobel, enfin… Med Sci (Paris) 2021; 37 : 77–80. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. La nanoparticule lipidique utilisée pour l’édition de gènes in vivo (extrait partiel et modifié de la figure 1 de [8]). |
| Dans le texte | |
|
Figure 2. Baisse progressive de la concentration sérique de la protéine TTR après infusion d’une seule dose de NTLA-1. Les doses employées (0,1 et 0,3 mg/kg) sont indiquées sur la figure ; les trois courbes de chaque panneau correspondent aux trois patients ayant reçu l’une ou l’autre dose de NTLA-1 (extrait partiel et modifié de la figure 4 de [8]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.