")
")
| Issue |
Med Sci (Paris)
Volume 37, Number 6-7, Juin-Juillet 2021
|
|
|---|---|---|
| Page(s) | 654 - 659 | |
| Section | Repères | |
| DOI | https://doi.org/10.1051/medsci/2021089 | |
| Published online | 28 June 2021 | |
CFTR et mucoviscidose, une histoire cinquantenaire
CFTR and cystic fibrosis, a 50-year long history
Laboratoire Signalisation et transports ioniques membranaires (STIM), université de Poitiers, 1 rue Georges-Bonnet, 86073, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
L’histoire médicale et scientifique de la mucoviscidose marquera durablement la médecine humaine. Il a fallu 50 ans d’expérimentations et d’observations médicales, favorisées par les progrès en génétique, en biologie moléculaire et en physiologie, pour construire la théorie ionique montrant que cette maladie est la conséquence d’un défaut généralisé du transport transépithélial de NaCl, et pour découvrir le gène responsable de la maladie. La découverte du gène CFTR et son clonage en 1989, la mise en évidence de ses mutations, puis la description de la protéine CFTR et de son rôle dans la maladie ont ainsi révolutionné la physiologie et la physiopathologie des transports ioniques des cellules épithéliales des systèmes respiratoire et digestif.
Abstract
The medical and scientific history of cystic fibrosis will have a lasting impact on human medicine. It will take 50 years of scientific experiments and medical observations, favored by advances in genetics, molecular biology and physiology to go from the ionic theory showing that this disease is the consequence of a generalized defect of the transepithelial transport of NaCl until the cloning of the CFTR gene in 1989. The discovery of the gene and its mutations, the description of the CFTR protein and its role in the disease have revolutionized the physiology and the pathophysiology of ionic transports in epithelial cells of the respiratory and digestive systems.
© 2021 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
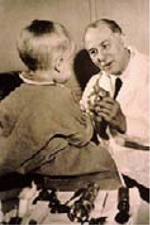
La mucoviscidose est, pour les populations d’origine européenne, la plus fréquente des maladies rares de transmission autosomique récessive, affectant principalement les poumons et plus généralement toutes les glandes sécrétoires exocrines. Elle est due à des mutations du gène CFTR (cystic fibrosis transmembrane conductance regulator), codant un canal qui permet l’échange d’ions chlorures entre l’intérieur et l’extérieur de la cellule. La frise historique présentée dans la Figure 1 montre les grandes étapes de la recherche sur la mucoviscidose, jusqu’à la découverte du gène CFTR, en 1989.
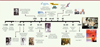 |
Figure 1. Frise historique représentant les principales découvertes scientifiques et médicales qui ont conduit à la découverte du gène CFTR. |
Vitamine A et fibrose kystique du pancréas
On connaît aujourd’hui cette maladie sous le nom de « mucoviscidose » ou, pour les anglophones, « cystic fibrosis » (CF, en français fibrose kystique). Mais les descriptions médicales des pédiatres pionniers de la première moitié du xx e siècle se concentrèrent d’abord sur le pancréas, avant d’élargir le diagnostic aux poumons.
Entre 1920 et 1933, les anatomo-pathologistes américains Kenneth D. Blackfan, Simeon B. Wolbach et Charles D. May furent intrigués par plusieurs cas d’autopsies d’enfants ayant des lésions histopathologiques du pancréas. Tous étaient carencés en vitamine A et sous-alimentés. Ces anatomo-pathologistes publient alors, entre 1933 et 1938, une série de notes pour décrire ces atteintes (obstruction, dilatation, atrophie et fibrose) touchant les glandes sécrétoires (en particulier les atteintes du pancréas et de la trachée et celles des glandes salivaires) [1, 2]. Ils proposent que ces lésions traduisent une seule et même maladie : tous ces patients, filles comme garçons, âgés de quelques semaines à 18 mois, décèdent de bronchopneumonie avec un pancréas exocrine très altéré. Blackfan et Wolbach vont alors apporter une première explication à ces observations pathologiques du pancréas ; ils écrivent : « nos études préliminaires indiquent que ces affections pancréatiques frappantes proviennent de la production anormale de sécrétions qui conduisent à la dilatation et à l’atrophie des canaux et acini » et proposent « qu’il est raisonnable de penser que ces lésions pancréatiques sont à l’origine d’un défaut de digestion des graisses et donc de la vitamine A » [1]. À la même époque, Guido Fanconi, professeur de pédiatrie à Zurich en Suisse, décrit précisément les modifications pathologiques des glandes pancréatiques et pulmonaires : « les changements pulmonaires et pancréatiques, deux organes vitaux, sont si profonds que leurs dérèglements apparaissent compréhensibles » [3]. Sydney Farber, pédiatre américain, note aussi que « les dommages du tractus respiratoire dépendent premièrement de leur obstruction par un mucus épais et d’un défaut d’une lubrification propre de l’épithélium cilié, et deuxièmement d’une infection à staphylocoque », mettant en avant l’atteinte pulmonaire et identifiant un défaut sécrétoire majeur : le mucus épais obstrue l’arbre bronchique, par un défaut de lubrification (hydratation) de l’épithélium cilié ; puis, secondairement à cette obstruction, une infection par la bactérie Staphylococcus aureus s’installe [4,5]. Charles May ira même jusqu’à penser que la rémission des patients (atteints de fibrose kystique du pancréas) dépend de la prévention et de la guérison de ces infections bactériennes pulmonaires [6]. Sydney Farber deviendra le pionnier de la chimiothérapie moderne. Il fera école auprès de Harry Shwachman, à Boston, qui, avec les pédiatres Paul di Sant’Agnese et Dorothy H. Andersen, à New York, se consacrèrent à la recherche sur la mucoviscidose. Dorothy Andersen établira finalement le lien entre les lésions néonatales du pancréas, les obstructions intestinales, la carence en vitamine A et les complications respiratoires. C’est à elle que l’on doit la reconnaissance de cette maladie en tant que seule et même entité [7], mais toujours liée au système digestif et, secondairement, à la fonction respiratoire.
Sur la piste du gène
Les travaux de Philip J Howard en 1944, Dorothy H Andersen et Richard G Hodges en 1946, puis de Charles Upton Lowe, Charles D May et Sheldon C Reed en 1949, posent les bases de l’hérédité mendélienne de la maladie [8-10]. L’analyse de Lowe, May et Reed est remarquable. Elle porte sur 134 enfants hospitalisés à l’hôpital de Boston entre 1938 et 1947 : 29 avec un iléus méconial1 et 105 avec une fibrose du pancréas. Ils émettent alors l’hypothèse que cette maladie, qui n’est pas fréquente dans la population, mais qui survient plusieurs fois dans une même famille, pourrait avoir une origine génétique [10]. Dans une famille, ils vont répertorier le nombre d’enfants, le rapprocher du nombre de ceux qui sont atteints de fibrose du pancréas, et déterminer si l’ordre de naissance est important. Ce dernier facteur ne montrera pas d’impact sur la survenue des naissances d’enfants malades, excluant ainsi une influence de l’âge de la mère lors de la procréation. Ils font également plusieurs observations importantes : aucun cas de fibrose du pancréas n’est diagnostiqué chez un adulte ; les enfants malades naissent de parents sains et décèdent avant la maturité ; les filles, comme les garçons, sont affectées. Ces résultats sont en accord avec ceux de Howard [8], Andersen et Hodges [9], et ne laissent dès lors plus de doute quant à l’origine génétique de la maladie pancréatique, de transmission récessive [10]. En comparant l’âge de décès des enfants et la survenue des lésions respiratoires, Lowe, May et Reed relient enfin précocité du décès et atteinte pulmonaire [10].
Les années cinquante fondatrices
Les années 1950 vont être riches en avancées et découvertes fondamentales sur plusieurs plans. Un nouvel acteur majeur de la recherche médicale va émerger dans plusieurs pays avec la création, entre 1955 et 1965, des fondations et associations de patients, parents et sympathisants (Figure 1), destinées notamment à récolter des fonds. Elles joueront un rôle majeur pour stimuler et financer la recherche, pour aider, conseiller et assister les patients et leur famille, et pour mobiliser les États et les entreprises du médicament contre cette maladie rare.
Le diagnostic de la mucoviscidose fait un bond spectaculaire à partir de l’été 1948, à New York, où une vague de chaleur intense survient le 28 août 1948. Paul di Sant’Agnese et Robert Darling admettent alors dans leurs services des enfants souffrant de déshydratation et de fibrose kystique du pancréas, dans un état de léthargie avancée. En comparant la composition de leur sueur à celle d’enfants non malades, ils vont faire une découverte importante en montrant une différence de concentration en sel (ion sodium Na+ et ion chlorure Cl-) dans la sueur sécrétée par les enfants malades, mais pas de différence dans la quantité de sueur. La concentration des deux ions se révèle en effet systématiquement plus élevée dans les prélèvements des enfants malades par rapport aux témoins. Au cours des cinq années qui vont suivre, Paul di Sant’Agnese va poursuivre cette analyse sur un nombre croissant d’enfants malades. Finalement, il établira une valeur seuil d’ions chlorure (de 60 milliéquivalents [mEq]/L) dans la sueur, au-delà de laquelle un diagnostic de la maladie doit être posé [11, 12]. Cette découverte va ouvrir une nouvelle ère de recherche pour la physiologie des glandes sécrétoires : l’analyse du contenu en électrolytes de la sueur (issue des sécrétions des glandes sudoripares situées dans la peau) permet de diagnostiquer une maladie…
La méthode sera améliorée par Lewis E. Gibson et Robert E. Cooke qui introduisent, en 1959, la stimulation locale sur l’avant-bras de l’enfant par la pilocarpine (un parasympathomimétique agissant sur les récepteurs muscariniques de l’acétylcholine) pour le recueil de sueur [13]. Ce test, appelé test de la sueur, fait désormais référence pour le diagnostic précoce (ou DNN, diagnostic néo-natal) de la mucoviscidose (→).
(→) Voir le numéro thématique La révolution médicale du dépistage néonatal, m/s n° 5, mai 2021
La malédiction populaire dite du « baiser salé sur le front de l’enfant malade » 2 du Moyen-Âge trouvait donc, trois siècles plus tard, une explication rationnelle et scientifique [14].
Dans une étude publiée en 1958, Harry Shwachman montrera qu’un diagnostic précoce, précis, avant l’âge de 3 mois, est fondamental pour obtenir un meilleur taux de survie des enfants atteints de « mucoviscidose » [15]. Mais à cette époque, le nom qu’il faut donner à cette maladie fait encore débat. Celui de « maladie fibrokystique du pancréas » (fibrocystic disease of the pancreas) n’est pas satisfaisant, car il exclut l’atteinte pulmonaire ou intestinale. Le nom de « mucoviscidosis » est alors proposé par Shwachman et Farber [4, 5] afin de se rapprocher de la théorie qui avançait comme explication une altération généralisée des glandes sécrétoires, une théorie défendue par eux-mêmes, mais aussi par Blackfan et Wolbach, entre 1933 et 1944. Cette dénomination ne sera cependant pas acceptée par tous, les glandes sudoripares ne produisant pas de mucus ! Finalement, issue de la dénomination « fibrose kystique du pancréas », sera conservée « fibrose kystique » (ou cystic fibrosis, en anglais) en délaissant la précision concernant le pancréas, car l’atteinte pulmonaire se révélait majeure dans la description clinique de la maladie.
Un second évènement important, mais indépendant, survient au Danemark avec les travaux du physiologiste Hans H Ussing. Celui-ci invente, en 1950, une méthode d’analyse des courants ioniques transépithéliaux [16]. Il va en effet, à partir de la peau de grenouille, modéliser les transports ioniques, identifiant ainsi une conductance Na+ apicale (coté muqueux) et une conductance K+ basolatérale (coté séreux). Cette méthode de Ussing est toujours utilisée pour étudier les transports ioniques transépithéliaux. Elle a permis de comprendre le rôle de la protéine CFTR et d’imaginer des molécules actives contre la mucoviscidose. Elle est fondée sur deux propriétés fondamentales des cellules épithéliales : leur polarité (un côté séreux, ou basolatéral, et un côté muqueux, ou apical) et leur étanchéité (cellules rendus jointives par des systèmes de jonctions intercellulaires). En utilisant deux couples d’électrodes mesurant le potentiel (exprimé en millivolts, mV) entre les deux faces d’un épithélium étanche, et en collectant le courant électrique qui le traverse (exprimé en microampères par unité de surface, µA/cm2), Hans Ussing déterminera le potentiel transépithélial (VTE), la résistance transépithéliale (RTE) et le courant transépithélial (dit courant de court-circuit). Le courant transépithélial résulte de l’activité des transporteurs, des récepteurs et des canaux ioniques qui sont répartis asymétriquement des deux côtés de l’épithélium.
Boite à outils et révolution moléculaire
Entre les années 1950 et 1980, la recherche sur la mucoviscidose va profiter des avancées conceptuelles et méthodologiques de la biologie et de l’électrophysiologie moléculaires (Figure 1). Les fondements de la biologie moléculaire émergent à partir des années 1940 et se poursuivent jusqu’aux années 1960, avec la description de l’ADN, de l’ARN, et de la synthèse des protéines. La génétique moléculaire va fournir les outils indispensables à l’analyse de l’ADN codant (par exemple, au début des années 1980, la PCR, ou réaction en chaîne par polymérase), le clonage par expression, le clonage par positionnement, le séquençage et l’expression des protéines par les méthodes de transfection hétérologue [17].
Parallèlement, l’électrophysiologie moléculaire, ou patch-clamp 3 [18, 19] (→), décrit les premiers canaux ioniques des cellules épithéliales [20]. Cette méthode révolutionnaire, inventée par Erwin Neher et Bert Sakmann, consiste en l’enregistrement de l’activité électrique de très faible intensité (de l’ordre du picoampère) qui traverse la membrane plasmique d’une cellule vivante [18]. Cette bioélectricité est le reflet du fonctionnement des canaux ioniques membranaires exprimés par la cellule. Elle permet d’enregistrer in situ leur activité, voire celle d’un seul canal ionique isolé, et d’en mesurer les variations physiologiques et pharmacologiques. C’est la méthode la plus précise pour mesurer en temps réel l’activité d’une protéine dans son environnement membranaire lipidique. Elle va guider la découverte du rôle fondamental des canaux ioniques en biologie et dans de nombreuses maladies (regroupées sous le nom de canalopathies) et transformera notre approche de la physiologie et de la médecine, ce qui vaudra à Neher et Sakmann le prix Nobel de physiologie ou médecine en 1991.
(→) Voir l’encadré de J. Teulon, m/s n° 5, mai 2004, page 550
La théorie ionique des glandes sudoripares, proposée par Paul di Sant Agnese en 1953 [11, 12], intrigue les physiologistes sur plusieurs points. Comment, en effet, relier deux faits en apparence contradictoires ? Les glandes sudoripares des patients fabriquent et secrètent un liquide trop riche en NaCl sans mucus, alors que le poumon et le pancréas exocrine élaborent un mucus déshydraté et visqueux. Comment le même défaut de transport ionique peut-il être à l’origine, dans un cas (les glandes sudoripares), d’une mauvaise réabsorption et, dans l’autre (poumons et pancréas), d’une sécrétion anormale ? Les travaux de Paul Quinton, de Michael R. Knowles et de Richard C. Boucher vont apporter des réponses. Paul Quinton, lui-même souffrant de mucoviscidose, sera le premier à montrer que le défaut fondamental dans cette maladie est lié à l’imperméabilité aux ions chlorure des cellules épithéliales des glandes sudoripares [21]. Physiologiquement, la sueur est fabriquée en deux étapes. D’abord, la glande produit un fluide primaire dont la concentration en NaCl est équivalente à celle du plasma. Ce fluide isotonique est ensuite véhiculé jusqu’à la surface de la peau par des canaux collecteurs. Au cours de ce transit, le NaCl est en partie réabsorbé vers le plasma. Le fluide résultant, dit secondaire, a donc une concentration en sel plus faible que le fluide primaire. Ce sont deux protéines, le canal sodique épithélial, sensible à l’amiloride (ENaC), et le canal CFTR, exprimés par les cellules canalaires sudoripares, qui règlent l’équilibre de réabsorption. L’épithélium canalaire sudoripare est donc de type absorptif. Dans la mucoviscidose, l’anomalie génétique et le défaut de fonctionnement de la protéine CFTR font que la sueur primaire n’est plus affinée. Elle se retrouve ainsi hyper-concentrée à la surface de la peau, expliquant le baiser salé et l’anomalie du test de la sueur. L’épithélium pulmonaire est, au contraire, de type sécréteur. Le canal CFTR est actif non plus dans le sens d’une absorption des ions chlorure vers le plasma, mais d’une sécrétion du plasma vers la surface de la glande, hydratant ainsi le mucus en surface. Le canal ENaC conduit les ions sodium de la lumière de la glande vers la cellule. Dans un poumon malade, l’absence de protéine CFTR fonctionnelle entraîne ainsi une hyperabsorption du sodium et un déficit d’hydratation du mucus, qui devient visqueux. La physiologie cellulaire explique donc les anomalies de fonctionnement inverses de ces deux tissus dans une même maladie.
Prenant appui sur cette nouvelle théorie, les recherches vont s’amplifier pour comprendre les bases du transport ionique épithélial et, grâce au patch-clamp, les premiers canaux ioniques de la membrane apicale des cellules épithéliales pulmonaires seront identifiés dès 1986 [20].
Deux autres approches expérimentales vont faire progresser l’analyse moléculaire des transports ioniques. Dans la première, la mesure de la différence de potentiel nasal (nasal potential difference ou NPD) permet d’estimer in vivo les conductions ioniques en fonction de la variation du potentiel transépithélial à travers l’épithélium de surface nasal, qui est accessible à une électrode externe de mesure reliée à un voltmètre. Michael Knowles et Richard Boucher mesurent cette valeur chez les patients et montrent que l’absorption de sodium est anormalement élevée et associée à une imperméabilité aux ions chlorure [22]. La perte de fonctionnalité du canal CFTR s’accompagne d’une perte de la conductance transmembranaire anionique, ce qui se traduit par une hyperpolarisation (potentiel plus négatif) du NPD, associée à une diminution globale de la conductance Cl-. Cette méthode de mesure fonctionnelle est très importante pour le diagnostic et l’évaluation thérapeutique de médicaments contre la mucoviscidose.
La seconde approche va appliquer la méthode de Ussing à des cultures primaires de cellules épithéliales pulmonaires [23]. En 1985, John H Widdicombe, Michael J Welsh et Walter E Finkbeiner mesurent les paramètres bioélectriques de cellules épithéliales isolées de trachées humaines, qui forment, en culture, un épithélium reconstitué, étanche et résistant. Leurs mesures révèlent qu’avec un épithélium provenant de patients atteints de mucoviscidose, le courant de court-circuit ne répond plus à des agonistes physiologiques qui, normalement, stimulent les transports transépithéliaux. Ils concluent alors que le défaut primaire chez ces patients est une diminution de la conductance Cl- apicale. En croisant ces différentes approches expérimentales, in vivo et in vitro, l’origine ionique de la mucoviscidose fut ainsi démontrée. Il ne restait plus qu’à découvrir le gène responsable.
Le Graal, enfin atteint
De 1985 à 1989, la course vers le gène s’accélère grâce à la génétique dite inverse (ou clonage par positionnement) [24] (→).
(→) Voir la Synthèse de M. Goossens, m/s n° 12, décembre 1991, page 1048
La recherche de liaisons génétiques, une stratégie qui permet d’identifier des régions du génome contenant un gène prédisposant à une maladie génétique, va permettre d’atteindre le but recherché, l’identification du gène responsable. La piste des marqueurs génétiques chez des patients atteints de mucoviscidose est suivie par plusieurs équipes, pour se rapprocher du locus du gène responsable [25]. En 1985, le bras long du chromosome 7 devient le meilleur candidat pour héberger ce gène, car deux marqueurs génétiques associés à la maladie s’y trouvent : le gène PON (qui code la paraoxonase ou apoprotéine K) et un fragment d’ADN nommé DOCRI-917 identifié par RFLP (restriction fragment length polymorphism)4 [24, 25]. Mais il faudra encore quatre années de « marche » et de « sauts » sur le chromosome 7 pour aboutir, en septembre 1989, à la publication des trois articles fondateurs [26-28]. Une délétion de trois paires de base (CTT), au niveau d’un gène, en positions 1 521 à 1 523 (c.1521_1523del), est identifiée dans plus de 70 % des cas de mucoviscidose. Elle se traduit, pour la protéine, par la perte de la phénylalanine en position 508 (mutation F508del ou p.Phe508del). La protéine est aussi décrite, elle est baptisée CFTR, pour cystic fibrosis transmembrane conductance regulator, pour rendre compte de son lien avec la maladie et de son rôle supposé dans la régulation des échanges ioniques membranaires [27]. Le gène identifié, CFTR (MIM#602421), contient 27 exons et s’étend sur 190 kilobases (kb) sur le bras long du chromosome 7 (locus 7q31.2). L’ARN messager (ARNm) de 6,5 kb est traduit en 1 480 acides aminés formant la protéine CFTR [26-28].
Conclusions
Les progrès et découvertes que nous avons décrits ont révolutionné la physiologie et la physiopathologie des glandes exocrines et de la mucoviscidose. Ils illustrent comment la connaissance du normal et du pathologique peuvent se nourrir l’une de l’autre. À la suite de la découverte du gène, naît le consortium international pour l’analyse génétique de la mucoviscidose (cystic fibrosis genetic analysis consortium) sous l’impulsion de l’association nord-américaine CFF (cystic fibrosis foundation) [29]. Le génotypage des patients enrichira les protocoles de diagnostic, en complément du test de la sueur. Il sera plus tard relié aux phénotypes des patients [30] (→).
(→) Voir la Synthèse de C. Férec et al., m/s, n° 5, mai 1994, page 631
La recherche d’une thérapie peut alors débuter. Elle va aboutir 30 ans plus tard aux premiers médicaments efficaces ciblant la protéine CFTR (→).
(→) Voir la Synthèse de C. Férec, page 618 de ce numéro
Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Occlusion intestinale du nouveau-né, due à l’arrêt du méconium anormalement épais dans l’iléon terminal.
« the baby who is kissed at birth and tastes salty will have a short life »
Le patch-clamp consiste à enregistrer l’activité électrique d’un fragment microscopique de membrane cellulaire, isolé électriquement du reste de la surface cellulaire et ne contenant que quelques canaux.
Les marqueurs moléculaires de type RFLP reposent sur la digestion d’un ADN cible par une ou plusieurs enzymes de restriction spécifiques des sites de restriction présents dans l’ADN. La longueur des fragments obtenus est caractéristique.
Références
- Blackfan KD, Wolbach SB. Vitamin A deficiency in infants, a clinical and pathological study. J Pediatr 1933 ; 3 : 679–706. [Google Scholar]
- Blackfan KD, May CD. Inspissation of secretion, dilation of the ducts and acini, atrophy and fibrosis of the pancreas in infants. J Pediatr 1938 ; 13 : 627–634. [Google Scholar]
- Fanconi G, Uehlinger E, Knauer C. Das coeliakiesyndrom bei angeborenerzysticher pankreasfibromatose und bronchiektasien. Wien Med Wochenschr 1936 ; 86 : 753–756. [Google Scholar]
- Farber S, Shwachman H, Maddock CL. Pancreatic function and disease in early life. I. Pancreatic enzyme activity and the celiac syndrome. J Clin Invest 1943 ; 20 : 827–833. [Google Scholar]
- Shwachman H, Patterson P, Farber S. Significance of altered viscosity of duodenal content in pancreatic fibrosis (mucoviscidosis). AMA Am J Dis Child 1950 ; 80 : 864–865. [PubMed] [Google Scholar]
- May CD. Fibrosis of the pancreas in infants and children. Proc R Soc Med 1944 ; 37 : 311–313. [PubMed] [Google Scholar]
- Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study. Am J Dis Child 1938 ; 56 : 344–399. [Google Scholar]
- Howard PJ. Familial character of fibrocystic disease of the pancreas. Am J Dis Child 1944 ; 68 : 330–332. [Google Scholar]
- Andersen DH, Hodges RC. Celiac syndrome V. Genetics of cystic fibrosis of the pancreas with consideration of the etiology. Am J Dis Child 1946 ; 72 : 62–80. [CrossRef] [PubMed] [Google Scholar]
- Lowe CU, May CD, Reed SC. Fibrosis of pancreas in infants and children: statistical study of clinical and hereditary features. Am J Dis Child 1949 ; 78 : 349–374. [CrossRef] [PubMed] [Google Scholar]
- Darling RC, di Sant’Agnese PA, Perera GA, Andersen DH. Electrolyte abnormalities of the sweat in fibrocystic disease of pancreas. Am J M Sc 1953; 225 : 67–70. [Google Scholar]
- Di Sant’Agnese PA, Darling RC, Perera GA, Shea E. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas. Pediatrics 1953 ; 12 : 549–563. [PubMed] [Google Scholar]
- Gibson LE, Cooke RE. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics 1959 ; 23 : 545–549. [PubMed] [Google Scholar]
- Taussig LM. Cystic fibrosis. New York: Thieme-Stratton, 1984 : 498 p [Google Scholar]
- Shwachman H, Kulczycki LL. Long-term study of one hundred five patients with cystic fibrosis. Studies made over a five- to fourteen-year period. AMA. Am J Dis Child 1958 ; 96 : 6–15. [Google Scholar]
- Ussing HH, Zerahn K. Active transport of sodium as the source of electric current in the short-circuited isolated frog skin. Acta Physiol Scand 1951 ; 23 : 110–127. [CrossRef] [PubMed] [Google Scholar]
- Kaplan JC, Delpech M. Biologie moléculaire et médecine, 3e ed. Collection De la biologie à la clinique. Paris : Flammarion Médecine-Sciences, 2007 : 820 p. [Google Scholar]
- Neher E, Sakmann B. Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature 1976 ; 260 : 799–802. [CrossRef] [PubMed] [Google Scholar]
- Teulon J. Le patch-clamp en bref. Med Sci (Paris) 2004 ; 20 : 550. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Welsh MJ. An apical-membrane chloride channel in human tracheal epithelium. Science 1986 ; 232 : 1648–1650. [CrossRef] [PubMed] [Google Scholar]
- Quinton PM. Chloride impermeability in cystic fibrosis. Nature 1983 ; 301 : 421–422. [CrossRef] [PubMed] [Google Scholar]
- Knowles MR, Gatzy JT, Boucher RC. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Eng J Med 1981 ; 305 : 1489–1495. [Google Scholar]
- Widdicombe JH, Welsh MJ, Finkbeiner WE. Cystic fibrosis decreases the apical membrane chloride permeability of monolayers cultured from cells of tracheal epithelium. Proc Natl Acad Sci USA 1985 ; 82 : 6167–6171. [Google Scholar]
- Goossens M. Biologie de la mucoviscidose : progrès récents et perspectives. Med Sci (Paris) 1991 ; 7 : 1048–1051. [Google Scholar]
- Tsui LC, Buchwald M, Barker D, et al. Cystic fibrosis locus defined by a genetically polymorphic DNA marker. Science 1985 ; 230 : 1054–1057. [CrossRef] [PubMed] [Google Scholar]
- Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989 ; 245 : 1059–1065. [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989 ; 245 : 1066–1073. [Google Scholar]
- Kerem BS, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989 ; 245 : 1073–1080. [PubMed] [Google Scholar]
- Tsui LC. Mutations and sequence variations detected in the cystic fibrosis transmembrane conductance regulator (CFTR) gene: a report from the cystic fibrosis genetic analysis consortium. Hum Mutat 1992 ; 1 : 197–203. [CrossRef] [PubMed] [Google Scholar]
- Férec C, Mercier B, Audrézet MP. Les mutations de la mucoviscidose : du génotype au phénotype. Med Sci (Paris) 1994 ; 10 : 631–639. [Google Scholar]
- Férec C. La mucoviscidose : du gène à la thérapeutique. Med Sci (Paris) 2021; 37 : 618–24. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Frise historique représentant les principales découvertes scientifiques et médicales qui ont conduit à la découverte du gène CFTR. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.