")
")
| Issue |
Med Sci (Paris)
Volume 37, Number 6-7, Juin-Juillet 2021
|
|
|---|---|---|
| Page(s) | 601 - 608 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2021090 | |
| Published online | 28 June 2021 | |
Le virus de la fièvre de la vallée du Rift et son étonnante protéine NSs
Rift Valley fever virus and the amazing NSs protein
1
CellNetworks, CIID (Cluster of Excellence and Center for Integrative Infectious Disease Research), Virology, University hospital Heidelberg, Im Neuenheimer Feld 344, 69120 Heidelberg, Allemagne
2
Univ. Lyon, INRAe, EPHE, IVPC (Infections virales et pathologie comparée), 50 avenue Tony Garnier, 69007 Lyon, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
**
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Le virus de la fièvre de la vallée du Rift (VFVR) est un agent pathogène transmis à l’homme et au bétail par la piqûre de moustiques. Ce virus, découvert au Kenya en 1930, est considéré par l’Organisation mondiale de la santé comme présentant un risque important de provoquer de vastes épidémies. Les moyens dédiés à la lutte contre le VFVR restent toutefois particulièrement limités et le virus est mal connu. Dans cette Synthèse, nous nous attacherons à présenter ce virus avant de nous intéresser plus spécifiquement à son facteur de virulence, la protéine NSs. Nous discuterons la capacité de cette protéine virale à former des fibrilles de type amyloïde et son implication dans la neurotoxicité du virus chez les animaux infectés.
Abstract
Rift Valley Fever Virus (RVFV) is an emerging zoonotic pathogen transmitted to humans and livestock through mosquito bites, which was first isolated in Kenya in 1930. The virus is classified by the WHO among the pathogens for which there is an urgent need to develop research, diagnostics, and therapies. However, the efforts developed to control the virus remain limited, and the virus is not well characterized. In this article, we will introduce RVFV and then focus on its virulence factor, the nonstructural protein NSs. We will mainly discuss the ability of this viral protein to form amyloid-like fibrils and its implication in the neurotoxicity associated with RVFV infection.
© 2021 médecine/sciences – Inserm
Vignette (Photo © Pierre-Yves Lozach).
Le virus de la fièvre de la vallée du Rift
Le virus de la fièvre de la vallée du Rift (VFVR) a été découvert par Robert Daubney et John Hudson en 1930, au Kenya, dans la vallée du Grand Rift, à l’est de l’Afrique [1]. Ce virus est pathogène pour l’homme et pour de nombreux animaux d’élevage, mais aussi sauvages [2]. Chez les animaux, et particulièrement chez les jeunes ruminants, l’infection se traduit par des malformations fœtales et des taux d’avortement et de mortalité importants [2]. Chez l’homme, les infections asymptomatiques et bégnines sont majoritaires. Néanmoins, 1 à 2 % des patients infectés développent des formes graves avec des affections durables, comme des rétinites, des hépatites, voire des fièvres hémorragiques et des encéphalites, pouvant nécessiter une hospitalisation [2]. Des flambées épidémiques ont éclaté au Kenya, en 1997-1998 et en 2006-2007, puis se sont propagées dans les pays voisins. Au cours de certaines épidémies, le taux de mortalité chez les personnes hospitalisées a atteint près de 40 % [2].
Le VFVR est essentiellement transmis aux animaux par des moustiques du genre Aedes et Culex, lors de leur repas sanguin [3]. Néanmoins, le plus souvent, l’homme contracte le virus par contact avec des liquides corporels ou des tissus infectés plutôt que par des piqûres de moustiques. Depuis sa découverte, le VFVR s’est propagé hors du continent africain pour atteindre l’île de Madagascar dans les années 1970, et plus récemment, l’Arabie Saoudite et la Turquie [2]. En plus du coût direct en termes de morbidité et de vies humaines perdues, les épidémies de VFVR ont eu des conséquences socio-économiques désastreuses [4]. L’épidémie de 2006 a, par exemple, coûté 471 millions de dollars (environ 392 millions d’euros) en productivité agricole à la Somalie, soit 5 % de son PIB (produit intérieur brut) [4].
Du fait de l’activité humaine et des changements climatiques, plusieurs espèces de moustiques impliquées dans la transmission du VFVR, dont Aedes albopictus, se sont implantées en Europe et leur expansion géographique ne cesse de progresser [5]. En 2017, l’Organisation mondiale de la santé (OMS) a classé le VFVR parmi les agents pathogènes les plus dangereux, susceptibles de provoquer de vastes épidémies. Les efforts de recherche et de prévention restent pourtant limités, et le virus est mal caractérisé. Dans cette Synthèse, nous présenterons le VFVR, son organisation structurale et son cycle cellulaire. Nous nous intéresserons ensuite à l’une des protéines du virus, la protéine NSs, et, particulièrement, à sa capacité à former des fibrilles de type amyloïde [6]. Les informations précises relatives à l’épidémiologie, aux manifestations cliniques et à l’impact socio-économique de l’infection par ce virus, pourront être consultées dans la revue de Daniel Wright et al. [2].
Organisation structurale des particules virales
Le VFVR est un virus du genre Phlebovirus (famille Phenuiviridae, ordre Bunyavirales). Les particules virales, globalement sphériques et de taille hétérogène, ayant un diamètre moyen de 100 nm [7], sont enveloppées. Elles comportent quatre protéines : la polymérase virale L et la nucléoprotéine N, localisées dans la particule, et les glycoprotéines d’enveloppe Gn et Gc, présentes à sa surface [8]. Ces deux dernières forment des protubérances à la surface des particules, responsables de leur attachement aux cellules hôtes (Figure 1). Le génome viral se présente sous la forme de trois segments d’ARN simple brin de polarité négative, dont les noms (S, M et L) se réfèrent à leur taille : L (large) pour le plus grand, M (medium) pour celui de taille intermédiaire et S (small) pour le plus petit. Chaque segment possède à ses extrémités 5’ et 3’ des séquences complémentaires conservées, qui s’apparient en de courtes structures double brin sur lesquelles se lie la polymérase virale L [9]. Il en résulte une pseudo-circularisation de chacun des segments qui, recouverts de la nucléoprotéine N, forment les ribonucléoprotéines. Ces structures sont souvent comparées à des épingles à cheveux (Figure 1). Le VFVR ne possède aucune structure interne rigide. La nucléoprotéine N est donc primordiale pour la protection du matériel génétique du virus.
 |
Figure 1. Organisation structurale des particules du VFVR. Les particules virales sont enveloppées d’une bicouche lipidique et recouvertes d’hétérodimères de glycoprotéines Gn et Gc. À l’intérieur, le génome tri-segmenté d’ARN simple brin est protégé par la nucléoprotéine N, qui, avec l’ARN polymérase ARN-dépendante (ApAd L), forme des ribonucléoprotéines pseudo-circulaires. Les trois segments génomiques viraux sont désignés en fonction de leur taille : S (petit), M (moyen) et L (grand). |
Entrée du virus dans la cellule hôte
L’infection débute par la fixation des virus aux cellules hôtes (Figure 2). Les héparanes sulfates présents à la surface des cellules1 facilitent l’infection par le VFVR, probablement en servant de molécules d’attachement [10, 11]. DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin) et L-SIGN (liver/lymph node-specific intercellular adhesion molecule-3-grabbing integrin), des lectines humaines de type C (dont les capacités de liaison aux ligands glucidiques sont dépendantes du calcium), constituent deux autres récepteurs d’entrée dans les cellules du VFVR [12-14] (→).
(→) Voir la Nouvelle de R. Meier et al., m/s n° 1, janvier 2012, page 16
 |
Figure 2. Cycle cellulaire du VFVR. Les particules virales s’attachent à la surface de la cellule par différents récepteurs, comme les héparanes sulfates, DC-SIGN ou L-SIGN (1) . Les particules sont ensuite internalisées dans la cellule (2) , avant de pénétrer dans le cytosol depuis les endosomes tardifs (3) . Après la libération du génome et du matériel viral dans la cellule, la réplication et la transcription de l’ARN génomique débutent (4) . Il s’ensuit l’expression des protéines virales (5) , puis l’assemblage de nouvelles particules au niveau de l’appareil de Golgi (6) . Les nouvelles particules, contenues dans des vésicules intracellulaires, gagnent alors la surface de la cellule (7) . Ces vésicules, après avoir fusionné avec la membrane plasmique, libèrent leur contenu à l’extérieur de la cellule (virions ou progénies virales) (8) . |
DC-SIGN est exprimée à la surface des cellules dendritiques localisées dans le derme, le site anatomique de transmission du virus. L-SIGN est présente à la surface de l’endothélium du foie ; la capacité de cette lectine à servir de récepteur du VFVR pourrait expliquer, au moins en partie, le tropisme hépatique du virus. Cependant, des tissus n’exprimant aucun de ces deux récepteurs sont permissifs à l’infection par le VFVR, indiquant que ce dernier peut utiliser d’autres mécanismes d’entrée. La multiplicité des récepteurs potentiels du VFVR contribue ainsi, probablement, à son très large tropisme tissulaire.
Après s’être attachées à la cellule, les particules virales sont internalisées dans les cellules par endocytose (Figure 2) [15-17]. Les vésicules contenant les virus fusionnent ensuite avec les endosomes tardifs dont l’acidité déclenche de multiples modifications conformationnelles des glycoprotéines d’enveloppe Gn et Gc [18]. La résolution des structures cristallines de Gn et Gc [19, 20] a récemment permis d’améliorer significativement notre compréhension des mécanismes de fusion du virus, révélant la capacité d’ancrage de ces protéines dans la membrane des endosomes. Ces modifications induites par le pH acide des endosomes tardifs déclenchent la fusion de l’enveloppe virale avec la membrane des endosomes. Cette étape de l’entrée du virus se produit environ vingt minutes après le début de l’internalisation des particules [15]. Un pore s’ouvre alors dans la membrane des endosomes et les ribonucléoprotéines virales sont libérées dans le cytosol.
Réplication et assemblage du virus
La réplication de l’ARN viral s’effectue exclusivement dans le cytosol de la cellule infectée. Le génome tri-segmenté du VFVR code au moins six protéines (Figure 3). Les segments L et M sont de polarité négative et codent, respectivement, l’ARN polymérase ARN-dépendante virale L (ou ApAd L) et un précurseur polypeptidique. Dans le réticulum endoplasmique, ce précurseur subit des clivages protéolytiques à l’origine de la formation des glycoprotéines d’enveloppe Gn et Gc, de la protéine non structurale NSm, et d’une glycoprotéine de 78 kDa (GP78). NSm favorise la propagation du virus en bloquant les mécanismes d’apoptose cellulaire déclenchés par l’infection de la cellule [21]. La GP78 aurait un rôle structural, mais uniquement dans les cellules des moustiques infectés [22]. Le segment S, quant à lui, code la protéine N et la protéine non structurale NSs, via un mécanisme de traduction ambisens2 (Figure 3). La réplication du virus ainsi que l’assemblage et la libération de nouvelles particules reposent sur des mécanismes complexes mal caractérisés. D’excellentes revues font la synthèse de l’état des connaissances les plus récentes sur ces sujets [2, 9]. Notons cependant que la polarité négative des segments d’ARN génomiques viraux empêche leur traduction directe par la machinerie cellulaire. Des ARN messagers (ARNm) intermédiaires sont donc nécessaires à la synthèse des protéines virales (Figure 3). Les ARNm viraux sont plus courts que les ARN génomiques et présentent une coiffe à leur extrémité 5’, suivie d’une brève séquence hétérogène dérivée de la cellule hôte ; par contre, ils ne possèdent pas de queue poly(A). Du fait de ses interactions avec les ARN viraux en cours de synthèse, la nucléoprotéine N joue un rôle important dans la réplication du virus, mais aussi dans l’incorporation de ces ARN dans les nouvelles particules virales [23]. Le virus bourgeonne au niveau des membranes de l’appareil de Golgi, où les particules virales acquièrent leur enveloppe lipidique et entrent dans la voie de sécrétion pour quitter la cellule (Figure 2) [23]. Une protéine résidente de l’appareil de Golgi, GBF1 (Golgi-specific brefeldin A-resistance guanine nucleotide exchange factor 1), impliquée dans la formation du complexe COPI (coat protein I) responsable du bourgeonnement des vésicules golgiennes, a récemment été impliquée dans l’assemblage et la sortie du virus [24].
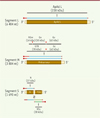 |
Figure 3. Organisation génomique du VFVR. Les ARNm viraux sont « coiffés » en 5’ et flanqués de courtes séquences hétérogènes dérivées de la cellule hôte (points et lignes rouges). Les ARNm transcrits à partir d’ARN génomiques sont représentés par les lignes en bleu clair. Le segment L code l’ARN polymérase ARN-dépendante L (ApAd L). Le segment M code le polypeptide précurseur, dont le clivage protéolytique produit la protéine non structurale NSm et les deux glycoprotéines d’enveloppe Gn et Gc. L’utilisation d’un site d’initiation de la traduction alternatif dans le segment M conduit à l’expression d’un second polypeptide précurseur, à l’origine d’une protéine de 78 kDa et de Gc. Le segment S code la nucléoprotéine N et la protéine non structurale NSs. L’ARNm de la protéine non structurale NSs (ligne verte) est synthétisé à partir d’ARN anti-génomique (ligne vert clair) (de 5’ vers 3’). Dans cette figure, les lignes noires symbolisent la traduction en protéines. nt : nucléotides. |
Réponse immunitaire contre le VFVR
L’immunité acquise contre le VFVR demeure peu documentée. La production d’anticorps neutralisant le virus apparaît toutefois comme une première ligne de défense, qui est induite chez tous les hôtes mammifères [2]. Les glycoprotéines de surface, Gn et Gc, en sont les cibles privilégiées. Des anticorps neutralisants bloquant le réarrangement fusogénique Gn-Gc en ciblant spécifiquement Gn ont ainsi été décrits [25]. Comme pour de nombreux microbes, parmi les premières cellules que rencontre le virus lors de sa pénétration dans le derme, les cellules dendritiques et les macrophages résidents, qui expriment notamment DC-SIGN, sont susceptibles d’être infectés [13, 26]. Mais la prolifération rapide des granulocytes neutrophiles ainsi que celle des lymphocytes B et des lymphocytes T CD4+ et CD8+, qui orchestrent la défense anti-virale, sont associées à des formes moins sévères de l’infection chez l’homme et les primates [2].
L’immunité innée joue un rôle essentiel dans la prévention de la maladie de la fièvre de la vallée du Rift [9]. Dans les modèles animaux d’infection par le virus, l’induction précoce de la réponse interféron (IFN) de type I est en effet corrélée au contrôle du virus et de sa propagation [9]. Les trois segments d’ARN du VFVR, dans leur conformation en épingle à cheveux, peuvent être détectés par RIG-I (retinoic acid-inducible gene I), une hélicase à ARN, responsable majeure de l’activation de la réponse IFN lors d’une infection. Le VFVR a cependant développé des stratégies pour retarder et se soustraire à cette réponse dans les cellules infectées, principalement grâce à sa protéine non structurale NSs [9].
NSs : un facteur de virulence du VFVR
NSs est une petite protéine virale de 30 kDa. Elle est exprimée dans les cellules infectées mais n’est pas incorporée dans les particules virales. Peu d’informations sont disponibles sur sa structure. Son étude est en effet difficile à réaliser : la forme complète de la protéine s’agrégeant facilement, il est difficile de la produire en solution. Seule une forme tronquée, dépourvue de l’essentiel de ses domaines N- et C-terminaux, soit environ la moitié de la protéine, a pu être analysée par diffraction des rayons X [27]. Les observations qui ont résulté de ces analyses ont été confirmées par des études de résonance magnétique nucléaire montrant que la protéine NSs présente des régions internes désordonnées et une forte propension de son domaine C-terminal à former des feuillets b [27, 28].
Dans les cellules infectées, NSs perturbe le transport nucléaire et les fonctions qui y sont associées [29, 30]. Son expression déstabilise également le cytosquelette d’actine de la cellule en ciblant l’un de ses régulateurs le plus important, la protéine Abl2 (ABL proto-oncogene 2) [31]. Il en résulte un profond changement morphologique des cellules infectées et une augmentation de leur mobilité cellulaire.
Outre ses effets sur les cellules infectées, NSs est également un puissant antagoniste de la réponse immunitaire innée. Cela a été montré par l’utilisation de l’isolat Clone 13, une souche naturelle de VFVR dépourvue du gène NSs. Cet isolat, contrairement à la souche sauvage, induit de fortes réponses IFN de type I et son infectivité pour le bétail et l’homme est fortement atténuée [9, 32]. Il en est de même chez les souris BALB/c infectées par cet isolat : ces souris survivent malgré la production et la propagation de virus infectieux [32], ce qui n’est pas le cas de souris inoculées avec le virus sauvage ; ces dernières développent une hépatite aiguë, non mortelle, mais qui est, en général, suivie d’une encéphalite fatale [32]. Du fait de son action inhibitrice sur la réponse immunitaire, NSs a été considérée comme un facteur de virulence majeur du VFVR chez les mammifères.
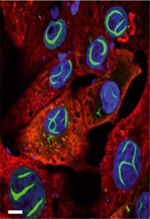
À côté de son rôle régulateur de la réponse immunitaire et de ses effets sur la cellule infectée, la protéine NSs est à l’origine de la formation de grandes structures filamenteuses dans le noyau des cellules, une caractéristique de l’infection par le VFVR (Figure 4A) [33]. L’ultrastructure et la fonction de ces filaments étaient jusqu’à présent très mal caractérisées, hormis leur implication dans des défauts de cohésion et de ségrégation des chromosomes [34]. Une étude récente, utilisant diverses techniques d’imagerie, a permis de révéler que NSs était capable de former des fibrilles de type amyloïde [6]. Cette étude a ainsi fourni une première description détaillée et novatrice de l’activité cellulaire de NSs. Elle a également confirmé la responsabilité de NSs dans la neurotoxicité observée chez les animaux infectés par le VFVR, associée à la formation de ces structures.
 |
Figure 4. La protéine non structurale NSs forme des faisceaux de fines fibrilles. A. Image de microscopie confocale de cellules Vero infectées par le VFVR, après marquage du noyau par le colorant Hoeschst (bleu) et par immunomarquage des protéines N (en rouge) et NSs (en vert). B. Micrographie électronique montrant un long filament nucléaire de NSs après immunomarquage par un anticorps dirigé contre NSs et conjugué avec de l’or colloïdal (points noirs). C. Image d’un filament nucléaire, comme celui présenté en B, à plus fort grossissement, révélant les fines fibrilles individuelles de NSs. L’or colloïdal (points noirs) est clairement visible. |
Des fibrilles de type amyloïde formées par NSs
Les fibrilles amyloïdes résultent d’un repliement et d’un assemblage aberrants de protéines en fibrilles linéaires ordonnées [35]. À ce jour, chez l’homme, une cinquantaine de protéines à l’origine de ces fibrilles et de ces dépôts tissulaires amyloïdes ont été identifiées. Ces dépôts peuvent être localisés dans certains tissus ou présents dans l’ensemble de l’organisme (systémiques) [35, 36] (→).
(→) Voir la Synthèse de G. Grateau et al., m/s n° 6-7, juin-juillet 2005, page 627
Beaucoup de ces fibrilles et de ces dépôts tissulaires sont associés à des maladies connues sous le nom d’amylose. Celles-ci sont souvent neurodégénératives, comme la maladie d’Alzheimer, la maladie de Parkinson et la maladie de Creutzfeldt-Jakob. Avec l’augmentation de l’espérance de vie, ces amyloses sont de plus en plus répandues dans le monde et, à ce jour, aucune n’est curable. Déterminer si l’agrégation des protéines amyloïdes est la cause ou une conséquence de la dérégulation de la machinerie cellulaire reste un défi scientifique. Les amyloses apparaissent généralement spontanément ou, moins souvent, par héritage génétique [37]. Des origines infectieuses ont également été évoquées mais, jusqu’à présent, les seules amyloses reconnues pour avoir une origine infectieuse étaient les maladies à prions [37]. Le VFVR, par ses capacités à induire ces agrégats, pourrait donc être considéré comme un nouveau déclencheur de ce type de maladies.
Le VFVR infecte un très grand nombre de types cellulaires. Pour élucider son rôle dans ces maladies, une dizaine de lignées cellulaires, chacune représentative de différents tissus et espèces, ont été testées. Toutes présentent, à la suite de l’infection, de larges filaments typiques de NSs dans leur noyau [6], et la seule expression de NSs par les cellules, en l’absence de toute autre protéine virale, conduit à la même formation de filaments nucléaires. L’analyse par microscopie électronique à transmission a permis de visualiser l’ultrastructure de ces filaments. Ils sont facilement reconnaissables par leur diamètre de 600 nm et leur longueur supérieure à 10 µm (Figure 4B). Les filaments produits par NSs sont, en fait, constitués de faisceaux de fines fibrilles de 12 nm de largeur, parallèles entre elles, une organisation structurale similaire à celle des dépôts amyloïdes (Figure 4C). Les agrégats amyloïdes ont en commun d’être résistants à la solubilisation par des détergents (tels que le sodium dodécylsulfate). C’est ce qu’a montré l’étude des agrégats de NSs, qui a nécessité, pour leur analyse, un recours à la technique de SDD-AGE (semi-denaturating detergent agarose gel electrophoresis), dédiée à l’étude des grands assemblages protéiques3 [6]. La nature amyloïde des fibrilles de NSs a également été confirmée par leur marquage avec la thioflavine-S (ThS), un colorant utilisé pour identifier et classer les agrégats amyloïdes [38].
Biogenèse des agrégats de NSs
Les agrégats amyloïdes se développent préférentiellement par l’incorporation continue de monomères de la protéine amyloïde, plutôt que par une augmentation du nombre de structures. Comme pour les agrégats amyloïdes, pour NSs, le nombre de filaments nucléaires reste stable, soit une demi-douzaine par noyau, et cela tout au long de l’infection. En revanche, leur taille s’accroît à mesure que la production de NSs augmente au cours de l’infection. Aux stades les plus tardifs de l’infection, la longueur des filaments de NSs peut ainsi atteindre 40 µm. Ces observations, confirmées par des analyses par imagerie sur cellules vivantes, ont par ailleurs permis de suivre la dynamique de formation des filaments de NSs en temps réel.
La polymérisation de certaines fibrilles amyloïdes dépend de mécanismes d’oxydo-réduction. NSs possède dans sa séquence protéique six résidus cystéine susceptibles de former par leur fonction thiol (-SH) des ponts disulfure (S-S) sensibles aux réactions d’oxydo-réduction. Le remplacement simultané des résidus cystéine, en position 39 et 40, par des résidus sérine, exempts de fonction thiol, abolit la virulence du VFVR chez les souris [39]. Ce double mutant NSs ne s’assemble plus en grands filaments, mais en petits amas sans forme, suggérant une corrélation entre les structures filamenteuses formées par NSs et la virulence du VFVR [6]. Un mutant similaire de NSs, qui n’induit pas l’apparition de larges filaments nucléaires mais conduit à la formation de courtes fibrilles, conserve en revanche sa virulence. Ces fibrilles correspondent probablement à des intermédiaires structuraux. Le processus d’oxydo-réduction semble donc avoir un rôle clef dans la formation des grands filaments de NSs. Néanmoins, les petites fibrilles intermédiaires semblent suffire à la virulence du VFVR.
Si NSs s’assemble en grands filaments nucléaires à des stades plus tardifs de l’infection, des agrégats sont également présents dans le cytosol des cellules infectées [6]. En microscopie à fluorescence, ces agrégats ont un aspect globulaire et sont 10 à 20 fois plus petits que les filaments que l’on observe dans le noyau. Leur ultrastructure rappelle celle des fibrilles d’un mutant de NSs (C149), qui sont très courtes, de l’ordre de quelques centaines de nanomètres. Il est probable que le potentiel d’oxydo-réduction dans le cytosol, à la différence de celui du noyau, ne permette pas l’assemblage des intermédiaires fibrillaires de NSs en de grandes structures filamenteuses. Quelle que soit leur apparence morphologique en microscopie, les agrégats de NSs sont constitués de fibrilles et leur ultrastructure est similaires à celle des dépôts amyloïdes.
Fonctions biologiques des fibrilles de NSs
NSs interfère avec la réponse immunitaire innée de diverses manières. Outre son effet sur la réponse IFN de type I, elle interfère en effet avec l’assemblage du facteur de transcription TFIIH (transcription factor IIH)4. Elle provoque ainsi l’arrêt général de la transcription des ARNm de la cellule infectée [40]. En plus de favoriser ainsi la synthèse des ARN du virus, il en résulte, entre autres, un blocage de l’expression des facteurs participant à la réponse immunitaire innée. Quelques heures seulement après le début de l’infection, NSs, en interagissant avec SAP30 (Sin3A-associated protein 30), réprime également l’expression du gène codant l’IFN-b [41]. Mais NSs aide aussi le virus à contourner les défenses de la cellule hôte en dégradant la protéine kinase R (PKR) [42]. Lors d’une infection, PKR, qui est activée par les ARN double brin, marque de la présence de complexes de réplication des ARN viraux. Ainsi activée, elle induit une cascade de signalisation qui conduit à l’inhibition de la synthèse de l’ensemble des protéines cellulaires. Ce mécanisme de protection de la cellule a pour but de prévenir la réplication et la propagation virale. La dégradation de PKR permet donc au VFVR de préserver l’intégrité de la machinerie cellulaire, essentielle à sa propre réplication et propagation.
La contribution des différentes structures de NSs au blocage de la réponse innée par le VFVR reste une question posée. En effet, le mutant de NSs qui s’assemble en petites fibrilles mais pas en larges faisceaux (voir plus haut) empêche toute transcription du gène codant l’IFN-b, et induit la dégradation de PKR aussi efficacement que la forme sauvage de la protéine [6]. En revanche, le mutant de NSs qui ne forme ni filaments ni fibrilles, mais des amas sans forme, en est incapable. La fibrillation de NSs, mais pas nécessairement la formation des grands filaments nucléaires, est donc critique pour l’échappement du VFVR à la réponse immunitaire innée. Récemment, il a été observé que des souris ne succombaient pas à l’infection par une souche du VFVR génétiquement modifiée afin d’exprimer un double mutant touchant les résidus lysine de la protéine NSs, mutant qui forme toujours des filaments, mais au profil d’expression très différent de celui du virus sauvage [43]. Davantage d’informations sur l’ultrastructure, le nombre et la dynamique de formation des filaments de ces mutants NSs dans le noyau et le cytosol des cellules infectées sont nécessaires. Cependant, ces résultats mettent en évidence le lien complexe entre le niveau d’infection, les diverses formes structurales de NSs et la virulence du VFVR.
Dépôts amyloïdes de NSs et neuro-virulence
La neuropathie est l’une des principales manifestations de l’infection par le VFVR [5]. Chez l’homme, elle pourrait donc se traduire par des atteintes de type amylose. Les souris BALB/c représentent un modèle intéressant de ces maladies. Ces souris, infectées par le VFVR, présentent en effet toutes les caractéristiques de l’évolution de la maladie souvent observée chez les personnes infectées : hépatite aiguë, précoce et non létale, suivie d’une encéphalite, tardive et mortelle. La présence de dépôts amyloïdes de NSs a été confirmée dans le cerveau de ces souris à la suite de l’infection [6]. Leur accumulation est particulièrement visible dans le tronc cérébral. La souche virale Clone 13 ne se propage pas jusqu’au cerveau des animaux, probablement en raison de l’incapacité de ce virus, dépourvu de NSs, à échapper à la réponse immunitaire innée. NSs apparaît donc, per se, un élément déterminant du neurotropisme du virus. L’inoculation intra-crâniale, que ce soit celle du virus sauvage ou celle de la souche Clone 13 à des souris, afin de favoriser le passage des virus dans le système nerveux central a ainsi conduit à des infections qui se sont révélées productives. Celles-là sont accompagnées d’une réplication efficace du virus et donc de la production de nouvelles particules virales infectieuses. Néanmoins, seules les souris exposées au virus sauvage présentent des dépôts de NSs de type amyloïde. Le fait que seuls ces animaux développent des paralysies et des convulsions, en plus de la présence des dépôts amyloïdes dans leur cerveau, indique que NSs peut être considérée comme le principal facteur viral responsable des troubles neurologiques associés à l’infection par le VFVR.
Perspectives
Les protéines amyloïdes peuvent être observées dans un large éventail d’hôtes, des bactéries à l’homme. Leur agrégation en fibrilles est souvent associée à des dysfonctionnements moléculaires et cellulaires, la plupart du temps à l’origine de maladies mortelles. Avec la protéine NSs, le concept de fibrilles amyloïdes a été étendu à d’authentiques infections virales, sans lien avec les protéines de l’hôte. De futures recherches devraient permettre de mieux appréhender les processus qui se cachent derrière la toxicité associée à la formation de ces substances amyloïdes virales, mais aussi les mécanismes de la formation de substances amyloïdes qui ont pour origine des protéines de l’hôte.
De nombreuses amyloses humaines, incurables et mortelles, sont de plus en plus répandues dans le monde. NSs apparaît donc comme un modèle de choix pour améliorer notre compréhension de l’agrégation des amyloïdes et de leur toxicité. Les fibrilles virales s’assemblent en effet rapidement, en seulement quatre heures ; elles sont aisément détectées, là où la majorité des fibrilles amyloïdes propres à l’hôte nécessitent généralement une activation artificielle, mettent des jours à se former, et sont difficiles à visualiser.
Les filaments de NSs sont la signature de l’infection par le VFVR. Une centaine d’autres virus appartiennent aux Bunyavirales, et la plupart codent une protéine NSs. Mais peu sont étudiés. Des virus non apparentés à ces virus, comme les polyomavirus et les adénovirus, codent également des protéines capables de former des structures filamenteuses [44], dont la caractérisation biochimique suggère qu’elles sont, pour certaines, des protéines amyloïdes [45, 46]5. Il est donc fort probable que d’autres virus produisent des dépôts amyloïdes. Déterminer le rôle exact de la formation de substances amyloïdes dans l’infection par ces virus reste donc un défi scientifique à relever.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Ce travail a été soutenu par des financements du CellNetworks Research Group (Université d’Heidelberg) et de la DFG (LO-2338/1-1 et LO-2338/3-1), ainsi que par des fonds INRAe, IDEX-Impulsion 2020 (Université de Lyon) et FINOVI (Fondation pour l’université de Lyon). Les micrographies électroniques présentées dans la Figure 4 sont une gracieuseté de Charlotta Fuyana (Plateforme de microscopie électronique, Université de Heidelberg). Nous remercions également Vibor Laketa et la plateforme d’imagerie du Centre de recherche intégrative sur les maladies infectieuses (Heidelberg).
Les héparanes sulfate sont des polysaccharides complexes appartenant à la famille des glycosaminoglycanes (GAG), présents à la surface des cellules et dans les matrices interstitielles.
La stratégie d’expression ambisens consiste en l’utilisation de deux cadres de lecture pour la synthèse des deux protéines, N et NSs, orientés de façon opposée, et séparés par une région intergénique dans le segment S. Un repliement structural spécifique de l’ARN dans la région intergénique est impliqué dans la terminaison de la traduction.
Dans cette technique, l’agarose est utilisé pour former le gel, remplaçant l’acrylamide, car les agrégats qui résistent aux détergents ont une taille comprise entre 200 et 4 000 kDa. Ils ne peuvent de ce fait entrer dans un gel de polyacrylamide conventionnel.
TFIIH est un complexe multiprotéique (10 protéines) impliqué dans la transcription de plusieurs gènes et la réparation de l’ADN.
Salladini E, Debarnot C, Delauzun V, et al. Phase transition and amyloid formation by a viral protein as an additional molecular mechanism of virus-induced cell toxicity. bioRxiv 2018. https://www.biorxiv.org/content/10.1101/497024v1
Références
- Daubney R, Hudson JR, Garnham PC. Enzootic hepatitis or Rift Valley fever. an undescribed virus disease of sheep cattle and man from East Africa. J Pathol Bacteriol 1931 ; 34 : 545–579. [CrossRef] [Google Scholar]
- Wright D, Kortekaas J, Bowden TA, Warimwe GM. Rift Valley fever: biology and epidemiology. J Gen Virol 2019 ; 100 : 1187–1199. [CrossRef] [PubMed] [Google Scholar]
- Lumley S, Horton DL, Hernandez-Triana LLM, et al. Rift Valley fever virus: strategies for maintenance, survival and vertical transmission in mosquitoes. J Gen Virol 2017 ; 98 : 875–887. [CrossRef] [PubMed] [Google Scholar]
- Peyre M, Chevalier V, Abdo-Salem S, et al. A systematic scoping study of the socio-economic impact of Rift Valley fever: research gaps and needs. Zoonoses Public Health 2015 ; 62 : 309–325. [CrossRef] [PubMed] [Google Scholar]
- Linthicum KJ, Britch SC, Anyamba A. Rift Valley fever: an emerging mosquito-borne disease. Annu Rev Entomol 2016 ; 61 : 395–415. [CrossRef] [PubMed] [Google Scholar]
- Leger P, Nachman E, Richter K, et al. NSs amyloid formation is associated with the virulence of Rift Valley fever virus in mice. Nat Commun 2020; 11 : 3281. [CrossRef] [PubMed] [Google Scholar]
- Freiberg AN, Sherman MB, Morais MC, et al. Three-dimensional organization of Rift Valley fever virus revealed by cryoelectron tomography. J Virol 2008 ; 82 : 10341–10348. [CrossRef] [PubMed] [Google Scholar]
- Léger P, Lozach PY. Bunyaviruses: from transmission by arthropods to virus entry into the mammalian host first-target cells. Future Virol 2015 ; 10 : 859–881. [CrossRef] [Google Scholar]
- Wuerth JD, Weber F. Phleboviruses and the type I interferon response. Viruses 2016 ; 8 : 174. [CrossRef] [Google Scholar]
- De Boer SM, Kortekaas J, de Haan CA, et al. Heparan sulfate facilitates Rift Valley fever virus entry into the cell. J Virol 2012 ; 86 : 13767–13771. [CrossRef] [PubMed] [Google Scholar]
- Riblett AM, Blomen VA, Jae LT, et al. A haploid genetic screen identifies heparan sulfate proteoglycans supporting Rift Valley fever virus infection. J Virol 2016 ; 90 : 1414–1423. [CrossRef] [PubMed] [Google Scholar]
- Leger P, Tetard M, Youness B, et al. Differential use of the C-type lectins L-SIGN and DC-SIGN for phlebovirus endocytosis. Traffic 2016 ; 17 : 639–656. [CrossRef] [PubMed] [Google Scholar]
- Lozach PY, Kuhbacher A, Meier R, et al. DC-SIGN as a receptor for phleboviruses. Cell Host Microbe 2011 ; 10 : 75–88. [CrossRef] [PubMed] [Google Scholar]
- Meier R, Helenius A, Lozach PY. DC-SIGN, un récepteur des phlébovirus : dynamique des interactions virus-récepteur. Med Sci (Paris) 2012 ; 28 : 16–18. [CrossRef] [EDP Sciences] [Google Scholar]
- de Boer SM, Kortekaas J, Spel L, et al. Acid-activated structural reorganization of the Rift Valley fever virus Gc fusion protein. J Virol 2012 ; 86 : 13642–13652. [CrossRef] [PubMed] [Google Scholar]
- Uckeley ZM, Koch J, Tischler ND, et al. Cell biology of phlebovirus entry. Virologie (Montrouge) 2019 ; 23 : 176–187. [Google Scholar]
- Albornoz A, Hoffmann AB, Lozach PY, Tischler ND. Early bunyavirus-host cell interactions. Viruses 2016 ; 8 : 143. [Google Scholar]
- Lozach PY, Mancini R, Bitto D, et al. Entry of bunyaviruses into mammalian cells. Cell Host Microbe 2010 ; 7 : 488–499. [CrossRef] [PubMed] [Google Scholar]
- Wu Y, Zhu Y, Gao F, et al. Structures of phlebovirus glycoprotein Gn and identification of a neutralizing antibody epitope. Proc Natl Acad Sci USA 2017 ; 114 : E7564–E7573. [CrossRef] [Google Scholar]
- Dessau M, Modis Y. Crystal structure of glycoprotein C from Rift Valley fever virus. Proc Natl Acad Sci USA 2013 ; 110 : 1696–1701. [CrossRef] [Google Scholar]
- Won S, Ikegami T, Peters CJ, Makino S. NSm and 78-kilodalton proteins of Rift Valley fever virus are nonessential for viral replication in cell culture. J Virol 2006 ; 80 : 8274–8278. [CrossRef] [PubMed] [Google Scholar]
- Kreher F, Tamietti C, Gommet C, et al. The Rift Valley fever accessory proteins NSm and P78/NSm-GN are distinct determinants of virus propagation in vertebrate and invertebrate hosts. Emerg Microbes Infect 2014 ; 3 : e71. [CrossRef] [PubMed] [Google Scholar]
- Hornak KE, Lanchy JM, Lodmell JS. RNA Encapsidation and packaging in the phleboviruses. Viruses 2016 ; 8 : 194. [CrossRef] [Google Scholar]
- Uckeley ZM, Moeller R, Kuhn LI, et al. Quantitative proteomics of Uukuniemi virus-host cell interactions reveals gbf1 as proviral host factor for phleboviruses. Mol Cell Proteomics 2019 ; 18 : 2401–2417. [CrossRef] [PubMed] [Google Scholar]
- Allen ER, Krumm SA, Raghwani J, et al. A Protective monoclonal antibody targets a site of vulnerability on the surface of Rift Valley fever virus. Cell Rep 2018 ; 25 : 3750–8 e4. [CrossRef] [Google Scholar]
- Terasaki K, Makino S. Interplay between the virus and host in Rift Valley fever pathogenesis. J Innate Immun 2015 ; 7 : 450–458. [CrossRef] [PubMed] [Google Scholar]
- Barski M, Brennan B, Miller OK, et al. Rift Valley fever phlebovirus NSs protein core domain structure suggests molecular basis for nuclear filaments. eLife 2017; 6 : e29236. [CrossRef] [PubMed] [Google Scholar]
- Cyr N, de la Fuente C, Lecoq L, et al. A OmegaXaV motif in the Rift Valley fever virus NSs protein is essential for degrading p62, forming nuclear filaments and virulence. Proc Natl Acad Sci USA 2015 ; 112 : 6021–6026. [CrossRef] [Google Scholar]
- Lau S, Weber F. Nuclear pore protein Nup98 is involved in replication of Rift Valley fever virus and nuclear import of virulence factor NSs. J Gen Virol 2020; 101 : 712–6. [CrossRef] [PubMed] [Google Scholar]
- Copeland AM, Van Deusen NM, Schmaljohn CS. Rift Valley fever virus NSS gene expression correlates with a defect in nuclear mRNA export. Virology 2015 ; 486 : 88–93. [CrossRef] [PubMed] [Google Scholar]
- Bamia A, Marcato V, Boissiere M, et al. The NSs protein encoded by the virulent strain of Rift Valley fever virus targets the expression of Abl2 and the actin cytoskeleton of the host, affecting cell mobility, cell shape, and cell-cell adhesion. J Virol 2020; 95 : e01768–20. [CrossRef] [PubMed] [Google Scholar]
- Bouloy M, Janzen C, Vialat P, et al. Genetic evidence for an interferon-antagonistic function of Rift Valley fever virus nonstructural protein NSs. J Virol 2001 ; 75 : 1371–1377. [CrossRef] [PubMed] [Google Scholar]
- Swanepoel R, Blackburn NK. Demonstration of nuclear immunofluorescence in Rift Valley fever infected cells. J Gen Virol 1977 ; 34 : 557–561. [CrossRef] [PubMed] [Google Scholar]
- Benferhat R, Josse T, Albaud B, et al. Large-scale chromatin immunoprecipitation with promoter sequence microarray analysis of the interaction of the NSs protein of Rift Valley fever virus with regulatory DNA regions of the host genome. J Virol 2012 ; 86 : 11333–11344. [CrossRef] [PubMed] [Google Scholar]
- Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 2014 ; 15 : 384–396. [CrossRef] [PubMed] [Google Scholar]
- Grateau G, Verine J, Delpech M, Ries M. Les amyloses, un modèle de maladie du repliement des protéines. Med Sci (Paris) 2005 ; 21 : 627–633. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Collinge J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 2016 ; 539 : 217–226. [CrossRef] [PubMed] [Google Scholar]
- Biancalana M, Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta 2010 ; 1804 : 1405–1412. [CrossRef] [PubMed] [Google Scholar]
- Monteiro GER, Jansen van Vuren P, Wichgers Schreur PJ, et al. Mutation of adjacent cysteine residues in the NSs protein of Rift Valley fever virus results in loss of virulence in mice. Virus Res 2018 ; 249 : 31–44. [CrossRef] [PubMed] [Google Scholar]
- Le May N, Dubaele S, Proietti De Santis L, et al. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell 2004 ; 116 : 541–550. [CrossRef] [PubMed] [Google Scholar]
- Le May N, Mansuroglu Z, Leger P, et al. A SAP30 complex inhibits IFN-beta expression in Rift Valley fever virus infected cells. PLoS Pathog 2008 ; 4 : e13. [CrossRef] [PubMed] [Google Scholar]
- Ikegami T, Narayanan K, Won S, et al. Rift Valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation. PLoS Pathog 2009 ; 5 : e1000287. [CrossRef] [PubMed] [Google Scholar]
- Li S, Zhu X, Guan Z, et al. NSs Filament formation is important but not sufficient for RVFV virulence in vivo. Viruses 2019 ; 11 : 834. [CrossRef] [Google Scholar]
- Erickson KD, Bouchet-Marquis C, Heiser K, et al. Virion assembly factories in the nucleus of polyomavirus-infected cells. PLoS Pathog 2012 ; 8 : e1002630. [CrossRef] [PubMed] [Google Scholar]
- McIntosh PB, Martin SR, Jackson DJ, et al. Structural analysis reveals an amyloid form of the human papillomavirus type 16 E1–E4 protein and provides a molecular basis for its accumulation. J Virol 2008 ; 82 : 8196–8203. [CrossRef] [PubMed] [Google Scholar]
- Pham CL, Shanmugam N, Strange M, et al. Viral M45 and necroptosis-associated proteins form heteromeric amyloid assemblies. EMBO Rep 2019 ; 20 : e46518. [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Organisation structurale des particules du VFVR. Les particules virales sont enveloppées d’une bicouche lipidique et recouvertes d’hétérodimères de glycoprotéines Gn et Gc. À l’intérieur, le génome tri-segmenté d’ARN simple brin est protégé par la nucléoprotéine N, qui, avec l’ARN polymérase ARN-dépendante (ApAd L), forme des ribonucléoprotéines pseudo-circulaires. Les trois segments génomiques viraux sont désignés en fonction de leur taille : S (petit), M (moyen) et L (grand). |
| Dans le texte | |
|
Figure 2. Cycle cellulaire du VFVR. Les particules virales s’attachent à la surface de la cellule par différents récepteurs, comme les héparanes sulfates, DC-SIGN ou L-SIGN (1) . Les particules sont ensuite internalisées dans la cellule (2) , avant de pénétrer dans le cytosol depuis les endosomes tardifs (3) . Après la libération du génome et du matériel viral dans la cellule, la réplication et la transcription de l’ARN génomique débutent (4) . Il s’ensuit l’expression des protéines virales (5) , puis l’assemblage de nouvelles particules au niveau de l’appareil de Golgi (6) . Les nouvelles particules, contenues dans des vésicules intracellulaires, gagnent alors la surface de la cellule (7) . Ces vésicules, après avoir fusionné avec la membrane plasmique, libèrent leur contenu à l’extérieur de la cellule (virions ou progénies virales) (8) . |
| Dans le texte | |
|
Figure 3. Organisation génomique du VFVR. Les ARNm viraux sont « coiffés » en 5’ et flanqués de courtes séquences hétérogènes dérivées de la cellule hôte (points et lignes rouges). Les ARNm transcrits à partir d’ARN génomiques sont représentés par les lignes en bleu clair. Le segment L code l’ARN polymérase ARN-dépendante L (ApAd L). Le segment M code le polypeptide précurseur, dont le clivage protéolytique produit la protéine non structurale NSm et les deux glycoprotéines d’enveloppe Gn et Gc. L’utilisation d’un site d’initiation de la traduction alternatif dans le segment M conduit à l’expression d’un second polypeptide précurseur, à l’origine d’une protéine de 78 kDa et de Gc. Le segment S code la nucléoprotéine N et la protéine non structurale NSs. L’ARNm de la protéine non structurale NSs (ligne verte) est synthétisé à partir d’ARN anti-génomique (ligne vert clair) (de 5’ vers 3’). Dans cette figure, les lignes noires symbolisent la traduction en protéines. nt : nucléotides. |
| Dans le texte | |
|
Figure 4. La protéine non structurale NSs forme des faisceaux de fines fibrilles. A. Image de microscopie confocale de cellules Vero infectées par le VFVR, après marquage du noyau par le colorant Hoeschst (bleu) et par immunomarquage des protéines N (en rouge) et NSs (en vert). B. Micrographie électronique montrant un long filament nucléaire de NSs après immunomarquage par un anticorps dirigé contre NSs et conjugué avec de l’or colloïdal (points noirs). C. Image d’un filament nucléaire, comme celui présenté en B, à plus fort grossissement, révélant les fines fibrilles individuelles de NSs. L’or colloïdal (points noirs) est clairement visible. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.