")
")
| Issue |
Med Sci (Paris)
Volume 36, Number 12, Décembre 2020
Vieillissement et mort : de la cellule à l’individu
|
|
|---|---|---|
| Page(s) | 1129 - 1134 | |
| Section | Mécanismes cellulaires et physiopathologie du vieillissement | |
| DOI | https://doi.org/10.1051/medsci/2020221 | |
| Published online | 09 December 2020 | |
Cause commune et mécanisme commun aux maladies du vieillissement ?
Common cause and mechanism for all pathologies of aging?
1
Mediterranean Institute for Life Sciences (MedILS), Meštrovic΄evo šetalište 45, 21000 Split, Croatie
2
Naos Institute for Life Sciences, 355 rue Pierre-Simon Laplace, 13593 Aix-en-Provence, France
3
Inserm U1001, Université Paris-Descartes, Faculté de médecine Paris-Descartes, 24 rue du faubourg Saint-Jacques, 75014 Paris, France
4
Center of Excellence for Science and Technology-Integration of Mediterranean Region (STIM-REI), Interdisciplinary Center for Advanced Sciences and Technology (ICAST), Université de Split, Poljička cesta 35, 21000 Split, Croatie
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
La santé est l’harmonie, le vieillissement et ses maladies la dysharmonie fonctionnelle aux niveaux moléculaire, cellulaire et tissulaire. Nos observations semblent suggérer une cause commune et un mécanisme commun du vieillissement et de ses nombreuses et diverses maladies. Cette cause commune est le dommage oxydatif de protéines particulières, résultant à la fois de leur mauvais repliement et du stress oxydatif. La cause commune va de pair avec l’horloge biologique des diverses maladies du vieillissement, dont l’incidence augmente exponentiellement avec l’âge, responsables de 90 % de la mortalité humaine. Des interventions pharmacologiques sur la cause commune pourraient éviter et atténuer simultanément toutes les maladies dégénératives et malignes, comme c’est le cas naturellement chez les super-centenaires.
Abstract
Health is harmony, aging and its diseases (are) functional disharmony at the molecular, cellular and tissue levels. Our observations lead us to think that there seems to be a common cause and a common mechanism for aging and its many and diverse diseases. This common cause is the oxidative damage to particular proteins emerging from a combination of imperfect folding and oxidative stress. This common cause jointly goes with the biological clock common to various age-related diseases, whose the incidence increases exponentially over time and causes 90% of human mortality. Pharmacological interventions on the common cause could avoid and simultaneously attenuate all degenerative and malignant diseases, as it is the natural case of super-centenarians.
© 2020 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.

Vignette (Photo © Inserm – Nissan Xavier/I-Stem).
Cette revue se veut synthétique et créative plutôt qu’exhaustive. Les revues sur la biologie cellulaire du vieillissement présentent souvent la description de détails moléculaires de mécanismes difficiles à comprendre par leur manque de concepts. Notre article propose que les concepts permettant de comprendre le vieillissement et les diverses et nombreuses maladies associées à l’âge sont plutôt simples et étiologiquement semblables. Ces concepts intègrent et interprètent les données existantes sans contredire significativement aucun résultat expérimental [1, 2].
Les protéines portent la fonction, et les gènes l’information biologique. Les protéines maintiennent la vie, les gènes assurent sa perpétuité/pérennité en gouvernant le renouvellement des protéines. Comme les protéines assurent toutes les fonctions de la vie – directement, ou indirectement en participant à la synthèse, à la réparation et au renouvellement des autres biomolécules –, la cause initiale (root cause) des maladies liées au vieillissement et du vieillissement même, doit se trouver au niveau du fonctionnement des protéines (Figure 1).
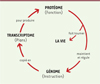 |
Figure 1. Interdépendance entre le protéome et le génome. L’ensemble des protéines (protéome) porte la fonction et assure le maintien de la vie. Dans le même temps, celles-ci assurent le maintien de l’ADN et la régulation de l’ensemble des gènes (génome) porteurs de l’information biologique. Le génome, quant à lui, assure la perpétuité et la pérennité de la vie, en maintenant le protéome par l’intermédiaire du transcriptome. Les protéines effectuent la synthèse, la réparation et le renouvellement des autres biomolécules. |
Cela a été démontré chez les bactéries, où l’endommagement sélectif des protéines au niveau cellulaire et en conditions physiologiques, provoque des changements phénotypiques et génétiques flagrants mais prévisibles [3].
On peut s’étonner qu’il y ait plus d’une centaine de théories du vieillissement [4] et autant de candidats à la cause du vieillissement et des maladies qui y sont associées : télomères, sirtuines, mitochondries et mitophagie, protéasome, autophagie, stress oxydant, mauvais repliement et agrégation des protéines et, généralement, la protéostase, etc. La recherche devient complexe si chacun de ces candidats est la cause du vieillissement. Mais si toutes ces causes sont les conséquences (ou les conséquences des conséquences…) d’une cause commune, il devient alors plus simple d’explorer la biologie du vieillissement et d’intervenir au plan thérapeuique.
Pour commencer nos études sur les phénomènes aussi complexes que la robustesse biologique, le vieillissement et les maladies dégénératives et malignes qui l’accompagnent, nous avons d’abord choisi les systèmes biologiques les plus simples. Notamment, les bactéries, la levure, le nématode et les rotifères [5, 6], avant de s’intéresser à l’homme. Si le vieillissement et la mort sont inhérents à la vie, on s’attend à trouver leurs traces au niveau moléculaire, déjà chez les organismes unicellulaires. Rappelons que la mort cellulaire programmée, ou apoptose, et son gène clé Tp53 (le plus fréquemment muté dans les cancers) ont été découverts chez un petit nématode (Caenorhabditis elegans) qui ne développe pas de cancers [7-9] !
Nos études comparatives sur Deinococcus radiodurans, une bactérie extrêmement résistante aux radiations, nous ont appris que la mort cellulaire est la conséquence de dégâts causés par l’oxydation des protéines, plutôt que de lésions radio-induites de l’ADN (Figure 2) [5, 10]. La question s’est alors posée de savoir si la radio-résistance et la chrono-résistance (résistance aux dommages induits au cours du temps) étaient liées… et la réponse a été oui. Ce dégât des protéines et leur agrégation pathologique augmentent exponentiellement (d’après la loi universelle de la mortalité, celle de Gompertz1) avec le vieillissement reproductif de la levure [1]. Nos études récentes sur les maladies de Parkinson [1] et de Charcot-Marie-Tooth (ou sclérose latérale amyotrophique, SLA) (non publiées) montrent que les effets des mutations qui prédisposent à ces maladies chez l’homme, prédisposent aussi la protéine mutée à l’oxydation par les espèces réactives de l’oxygène (ERO, ou reactive oxygene species [ROS] en anglais). Cela nous permet de proposer un mécanisme moléculaire et cellulaire du vieillissement et des maladies liées à l’âge (Figure 3) qui intègre et interprète la littérature sur ces sujets.
 |
Figure 2. Corrélation entre mortalité cellulaire et carbonylation des protéines. Cette corrélation est observée pour deux types de bactéries : Escherichia coli (Eco) utilisée comme modèle, et Deinococcus radiodurans (Dra) qui est extrêmement résistante aux rayonnements (UV et g), utilisés pour induire la carbonylation des protéines et la mort. Pour deux espèces animales, le rotifère bdelloïde Adineta vaga qui est radiorésistant, et le ver modèle Caenorhabditis elegans, la mortalité est celles des cellules germinales (stérilité) (figure adaptée de [10]). |
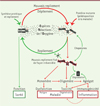 |
Figure 3. Mauvais repliement et oxydation des protéines à l’origine des maladies liées au vieillissement. La protéine mal repliée ou mutante est plus sensible aux attaques par les espèces réactives de l’oxygène (ERO) et sera donc oxydée. Deux processus concurrents vont entrer en jeu : soit cette modification est réversible, et la protéine sera repliée dans sa conformation native par l’action des protéines chaperonnes, maintenant ainsi sa fonction (flèches vertes) ; soit la modification (carbonylation) fixe sa mauvaise conformation, qui n’est alors plus sujette à l’action des protéines chaperonnes (flèches rouges). La protéine oxydée et mal repliée peut alors suivre différentes voies : 1) soit elle est directement dégradée par les protéases ; 2) soit elle évite le système de dégradation de la cellule et reste sous forme de monomère dysfonctionnel, conduisant à la maladie (le cancer par exemple) ; 3) soit elle forme des structures multimériques (oligomères et agrégats) toxiques pour la cellule, à l’origine, entre autres, de l’apparition de maladies neurodégénératives. La présence d’agrégats immunogéniques induit une réponse du système immunitaire qui enclenche l’apparition et le développement des maladies liées au vieillissement. |
La biologie du vieillissement
La biologie du vieillissement a fait l’objet de nombreuses revues exhaustives [2, 4, 11], et il serait redondant d’en refaire une ici. Le vieillissement est une dégradation progressive des fonctions biologiques qui engendrent plusieurs conditions (telles l’arthrite), maladies (cardiovasculaires, neurodégénératives et malignes) et la mort. Les manifestations du vieillissement nous démontrent la finesse de l’harmonie (homéostasie) chimique, moléculaire, cellulaire et tissulaire, d’un organisme jeune et sain. Apparemment, le fonctionnement harmonieux des organes assure la santé pendant des décennies, malgré les stress fréquents de la vie et ceux engendrés par nos habitudes.
Le vieillissement progressif induit un bruit diffus dans l’harmonie biochimique de l’organisme, qui s’accompagne de l’augmentation du risque de maladies et de mort. Nous proposons ci-dessous une nature chimique de ce bruit. Il existe un polymorphisme prononcé des phénotypes du vieillissement et de l’âge auquel ils apparaissent. Si l’on considère la longévité et les maladies liées à l’âge, les différences interindividuelles sont encore plus remarquables. Les différences de longévité entre les espèces animales s’étalent dans le temps, séparées d’un facteur dix-mille [12]. Et pourtant, le vieillissement et les maladies ne sont donc pas une fatalité, mais des processus biologiques complexes et flexibles, qui ont été manipulés spectaculairement au cours de l’évolution. Peut-on les manipuler aujourd’hui chez l’homme ?
Les maladies dégénératives et malignes liées à l’âge
On estime que le nombre de maladies dégénératives et malignes associées à l’âge est de l’ordre d’une centaine dans chaque catégorie. Les maladies dégénératives typiques sont, par exemple, l’arthrite rhumatoïde, l’ostéoporose, les maladies cardio-vasculaires, neuro-dégénératives et immunitaires ; elles sont toutes associées à des inflammations chroniques. Environ 90 % des cancers sont des carcinomes (cancers de tissus solides), dont l’incidence augmente avec l’âge, comme celle des maladies dégénératives. La dégénérescence du système immunitaire, au cours du vieillissement, fait que l’on doit aussi inclure une panoplie de maladies infectieuses, telles que la pneumonie, la COVID-19 et la grippe, comme étant des maladies liées à l’âge.
L’ensemble de ces deux à trois cents maladies liées à l’âge sont la cause d’environ 90 % de la mortalité humaine dans les pays économiquement développés. On s’approche progressivement de ce chiffre dans les pays en voie de développement.
Le dysfonctionnement cellulaire peut engendrer une perte de la masse cellulaire à l’origine des maladies dégénératives, soit un excès pathologique de cellules résultant d’un dérèglement de la division cellulaire qui caractérise les cancers. Or, le point commun de ces centaines de pathologies si diverses est que leur fréquence et leur taux de mortalité augmente exponentiellement avec l’âge. Ces diverses maladies ont donc une « horloge biologique » très semblable. Une cause commune de toutes les maladies liées à l’âge expliquerait ainsi facilement leur horloge biologique commune. Nous examinerons dans cette revue, l’identité de la cause commune aux maladies du vieillissement.
Les maladies comme expressions phénotypiques des défauts cellulaires
Les difficultés de guérison des maladies liées à l’âge, comme les cancers, sont probablement dues à la stabilité des altérations génomiques somatiques acquises aléatoirement dans les cellules individuelles qui en sont leurs causes, telles les mutations ou la méthylation de l’ADN. D’où l’intérêt des technologies de correction de mutations in vivo. Vu la précision toujours limitée de ces technologies par rapport au grand nombre des cellules touchées par les changements génétiques et épigénétiques et à leur localisation incertaine, le succès de guérisons des maladies par ces technologies reste néanmoins incertain.
En revanche, la manipulation du phénotype engendré par ces altérations génomiques paraît plus prometteuse. La maladie est un phénotype pathologique, une conséquence fonctionnelle de défauts génétiques et épigénétiques. Pour la santé, nous sommes concernés par le phénotype, pas par le génotype (d’autant plus qu’il reste souvent « silencieux » par rapport aux maladies). Notre génome est truffé de mutations silencieuses, ce que l’on appelle le polymorphisme – environ 30 000 différences par substitution d’acides aminés sont observées entre les protéomes de deux personnes. Une fraction inconnue de ces mutations est sans conséquence – phénotypiquement silencieuses – grâce à l’activité des protéines chaperonnes [13] qui atténuent le défaut fonctionnel de la protéine. En effet, pour ces protéines, le changement d’acides aminés liés à la mutation n’a pas d’impact direct sur son activité, mais peut affecter sa stabilité et son oxydabilité au cours du vieillissement.
De longues périodes de latence (ou « silence phénotypique ») sont observées pour des maladies qui ont été initiées à l’origine ou au cours de la vie. Ce silence peut reposer sur une compensation de la dysfonction cellulaire qui engendre la maladie, que l’on a nommée parabiose cellulaire [2, 14]. La parabiose cellulaire est une complémentation fonctionnelle trans-cellulaire. C’est une sorte de « solidarité cellulaire », avec échange et redistribution de « biens » entre cellules en contact, via diverses structures permettant le trafic moléculaire entre les cellules [2, 15, 16]. C’est ce qu’on appelle l’homéostasie tissulaire : la mise en commun des activités cellulaires, en transgressant les membranes cellulaires. C’est un mécanisme puissant de robustesse cellulaire. La suppression phénotypique de mutations récessives a en effet été démontrée et reconstituée en culture cellulaire [2], de même que la complémentation d’un défaut mitochondrial (respiration cellulaire), par le transfert des mitochondries entières à partir des cellules normales via les TNT (tunneling nanotubes) [17, 18]… L’inflammation chronique empêche la parabiose cellulaire. Elle isole les cellules in situ par destruction des connections intercellulaires [2]. Or, les inflammations dites « stériles » (sans infection), et leurs biomarqueurs, augmentent avec l’âge, et activent les phénotypes cellulaires pathologiques latents. Ceci est bien connu des cliniciens et brillamment étudié en cancérogénèse expérimentale sur la peau des souris [2]. Après les bombardements atomiques d’Hiroshima et de Nagasaki, au Japon, en 1945, il a fallu attendre 20 à 25 ans pour voir apparaître des cancers solides induits, mais dix fois moins de temps pour les leucémies, malignité des cellules circulantes qui ne sont pas sujettes à la parabiose cellulaire.
Génome et protéome : l’œuf et la poule…
Une des théories du vieillissement parmi les plus attractives est la théorie de la catastrophe d’erreurs de Leslie Orgel [19, 20]. Elle dit, essentiellement, que le taux d’erreur dans la synthèse des protéines étant au moins cent mille fois plus élevé que celui de la réplication de l’ADN, la fraction portant un acide aminé erroné est d’environ 10 % sur l’ensemble des protéines. S’il s’agit d’une ADN polymérase, elle sera à l’origine de plus d’erreurs dans la réplication de l’ADN, et donc générera plus de mutations [3]. Or, plus de mutations engendrent plus de mauvaises protéines… Et plus de mauvaises protéines, plus de mauvais ADN, et ainsi de suite. S’établiront ainsi des cercles vicieux qui se traduiront en vieillissant par un enchaînement catastrophique d’erreurs [19].
En considérant les prémisses d’Orgel, nous proposons que les dégâts chimiques des protéines, notamment l’oxydation, s’ajoutent aux taux d’erreurs de traduction déjà élevés, avec des effets semblables ou plus sévères que les erreurs de synthèse (Figure 1). La manipulation sélective des taux d’oxydation des protéines (sans changer le niveau d’ERO qui en sont responsables !) donne des effets mutagènes beaucoup plus puissants que les lésions de l’ADN [3] ; le paradigme a ainsi été modifié [1] (Figure 1).
La relation entre protéine et ADN ressemble au paradigme de l’œuf et de la poule, où le protéome est la poule et l’ADN, l’œuf : rien ne peut être plus mutagène que la modification du protéome dédié à l’ADN, à sa réparation et sa réplication. Le taux des mutations spontanées augmente exponentiellement avec les dégâts oxydatifs irréparables (comme la carbonylation2) des protéines erronées par les mutations [1,3].
Il est révélateur que tout changement phénotypique est possible par la modification directe et toxique des protéines qui y sont associées. Cela suffit pour créer les maladies. Dans ce cas, celles-ci, ainsi que les phénotypes induits, doivent donc être réversibles, en diminuant le niveau de protéines endommagées – à l’exception des conséquences stables, telles que les mutations ou modifications épigénétiques de l’ADN [1].
Mauvais repliement et oxydation des protéines
La structure des protéines leur a permis de développer, évolutivement, une résistance à l’oxydation qui protège ainsi leur fonction. Mais, dans leurs conformations natives, leur résistance aux dommages oxydatifs reste fragile. En effet, cette résistance est perdue du fait de leur mauvais repliement, provoquant leur oxydation pouvant mener à la perte de leur fonction. L’augmentation du taux d’erreur dans la biosynthèse des protéines (par mutations ribosomales ou par action d’antibiotiques qui agissent sur les ribosomes), ou dans leur repliement (par mutations des protéines chaperonnes), augmente proportionnellement leurs niveaux d’oxydation [1,3]. Réciproquement, la réduction de ces erreurs réduit leur oxydation et les effets phénotypiques [1,3].
L’oxydation punit les protéines imparfaites ! Mais, dans les cellules, plusieurs systèmes de défense protègent les protéines de l’oxydation : des molécules antioxydantes (acide ascorbique, polyphénols, etc.) ; des enzymes qui éliminent directement les oxydants (superoxyde dismutase, etc.) ; des systèmes enzymatiques de réparation des dommages (méthionine sulfoxyde réductase, protéine disulfure réductases, etc.) ; et des mécanismes d’élimination des protéines endommagées (protéasomes, lysosomes, protéases, etc.). En dépit de cette multitude de mécanismes de défense, une augmentation de la quantité de dommages oxydatifs des protéines est observée avec le vieillissement [21,22]. Ceci peut être dû à une augmentation de la formation d’oxydants ou de leur exposition, à une diminution ou une défaillance des systèmes de défense, ou un effet combiné des deux. Les protéines imparfaitement structurées sont donc la cible de deux activités antagonistes qui entrent en compétition : (1) les activités des protéines chaperonnes qui les replient et les rendent résistantes à l’oxydation, et (2) les activités non-enzymatiques d’oxydation, qui les rendent non repliables par les protéines chaperonnes et figées dans leur mauvaise conformation (Figure 3).
Les protéines oxydées : la cause du vieillissement et des maladies
Chez les bactéries, la perte de fonction ou une fonction altérée provoquée par un mauvais repliement, dépend d’une oxydation irréversible de la protéine, comme la carbonylation [3,10]. Le fait que l’activation des protéines chaperonnes par un mauvais repliement des protéines dépende de l’oxydation signifie qu’elles restent liées aux protéines mal repliées sans en pouvoir corriger la structure. Or, à l’état mal replié, les protéines ont certains de leurs acides aminés hydrophobes exposés à leur surface, dépliant davantage leur structure, et s’exposant davantage aux ERO qui pourront modifier un acide aminé exposé sur la chaîne latérale, ce qui conduit à une modification de la charge de la protéine. Cela affectera sa stabilité, sa conformation et donc sa fonction [23]. À proximité, les zones hydrophobes de la protéine auront tendance à se rapprocher, en chassant les molécules d’eau. Ainsi commence l’agrégation des protéines oxydées et mal repliées.
Suite à une insuffisance du système de nettoyage, l’excès de carbonylation est responsable de l’agrégation de protéines. Castro et al. ont montré que lorsque la carbonylation précède l’agrégation, un environnement oxydatif mène à l’agrégation de l’actine dans des lymphocytes T, et affecte la fonction de ces cellules [24]. Tanase et al. ont observé, suite à l’induction d’un stress oxydatif produisant des protéines carbonylées chez de jeunes souris, l’apparition d’agrégats identiques à ceux accumulés chez des souris âgées. Ces observations supportent ainsi l’idée que les modifications des protéines associées à un stress oxydatif s’accumulent avec le vieillissement et sont responsables de l’augmentation de l’agrégation des protéines et de l’altération de la protéostase [25]. Rahim et al. ont montré, en étudiant plusieurs centaines de protéines, qu’une interaction entre deux carbonyles provenant d’une même protéine, pouvait influer sur leur structure. Cette observation vient également supporter la notion que l’oxydation peut précéder la dénaturation et l’agrégation des protéines par réticulation [26].
D’autres voies peuvent influencer l’oxydation des protéines et leur agrégation, notamment la modulation de l’activité des protéines chaperonnes, en modifiant l’interaction entre protéines partenaires [27], ou en altérant la capacité de fonctionnement du protéasome et de l’autophagie [28].
Quand plusieurs protéines oxydées se stabilisent et forment une structure triangulaire ou circulaire (comme un pore), le processus d’agrégation doit recommencer et une nouvelle nucléation formera la prochaine structure oligomérique, etc. Si les oligomères gardent une structure qui n’est pas fermée, ou si les oligomères commencent à fusionner, la « polymérisation » peut se poursuivre et former de grands agrégats intra- et extracellulaires comme ceux observés dans les tissus affectés par la majorité des maladies dégénératives, notamment neurodégénératives.
Les paramètres intrinsèques de l’agrégation des protéines ne sont pas complétement compris, mais le fait que certaines protéines soient plus enclines à s’agréger est connu depuis longtemps : des peptides poly Q, dans la maladie d’Huntington ; des fibrilles b-amyloïde (Aβ), dans la maladie d’Alzheimer, par exemple. La plupart des protéines possèdent une ou plusieurs régions prédisposées à l’agrégation (ou en anglais, APR, aggregation prone regions). Mais celles-ci sont protégées par des interactions protéiques ou par des caractéristiques structurales spécifiques (enfouies dans le cœur hydrophobe de la protéine). Trois structures anormales des protéines oxydées qui sont de taille ont des effets biologiques différents et distincts (Figure 3) :
1) Les monomères oxydés perdent leur fonction ou fonctionnent mal, ce qui peut provoquer l’apparition de divers phénotypes pathologiques. Selon leur fonction initiale et dépendant du niveau cellulaire de ces monomères protéiques oxydés, les changements phénotypiques vont émerger [1, 3]. Ces nouveaux phénotypes sont en principe réversibles grâce au renouvellement des protéines. Par exemple, chez les bactéries, à quantité d’ERO constante, l’oxydation exclusive de leurs protéomes génère des phénotypes réversibles et variés qui dépendent de l’importance des dommages des protéines [3]. La carbonylation du protéome s’accompagne d’une augmentation exponentielle du taux de mutations spontanées [3], avec pour conséquence possible l’initiation de cancers. En effet, plus de protéines oxydées et d’agrégats sont retrouvés dans les cellules cancéreuses. Ainsi, il a été proposé que certains cancers pouvaient être considérés comme des maladies associées à l’agrégation des protéines [29].
2) Les oligomères sont des structures circulaires constituées de protéines mal repliées et oxydées, qui ressemblent aux pores hydrophobes. Ce sont les structures les plus cytotoxiques, qui s’insèrent et/ou se forment au sein des zones lipophiles des membranes. Ils sont ainsi responsables de la perte d’intégrité des membranes intracellulaires (mitochondriales, réticulum endoplasmique, et autres) et cellulaires (membrane plasmique). Cette cytotoxicité a été démontrée pour les oligomères d’α-synucléine, de superoxyde dismutase (SOD-1), de peptide β-amyloïde et d’amyline, contre des cellules humaines (dont les neurones) et contre les bactéries où ils ont un rôle antibiotique [1, 30, 31]. Les oligomères cellulaires formés par les protéines oxydées et mal repliées agissent comme des armes « faites maison » pour le suicide cellulaire. Ils apparaissent ainsi comme les meilleurs candidats de la cause initiale des maladies dégénératives (neurodégénératives, diabète de type 2, sarcopénie, etc.).
3) Les grands agrégats amyloïdes et fibrinoïdes sont généralement extracellulaires et moins cytotoxiques que les oligomères. Ils peuvent induire une réponse du système immunitaire (apparaissant comme du « non-soi ») qui pourrait être à l’origine des maladies auto-immunes. Un grand nombre des maladies auto-immunes liées à l’âge existe, comme l’arthrite rhumatoïde, le diabète, le psoriasis, et plus généralement, les maladies associées à une inflammation chronique, qui est suffisante pour que les agrégats deviennent pathogènes au niveau tissulaire. En réduisant la parabiose cellulaire, l’inflammation accélère en effet l’expression phénotypique (la manifestation) de la maladie [2] (Figure 3).
Le scénario qui permet d’expliquer la quasi-totalité des maladies liées à l’âge (Figure 3) est relativement simple. Il englobe et interprète tous les aspects du vieillissement et l’ensemble des maladies qui y sont liées. Par exemple, la télomérase, l’enzyme qui ajoute les répétitions d’ADN télomérique aux extrémités des chromosomes, est très peu abondante, mais elle est parmi les protéines les plus sensibles à la carbonylation [23]. On s’attend donc à ce que le vieillissement et le stress oxydant réduisent son activité, provoquant ainsi le raccourcissement des télomères et la sénescence cellulaire.
L’aspect le plus intéressant du modèle présenté dans la Figure 3 , est qu’avec un bouclier protecteur pharmacologique contre l’oxydation des protéines, il est possible de prévenir et/ou d’atténuer la quasi-totalité des maladies dégénératives et malignes liées à l’âge. Cela signifie qu’il est possible d’influencer le futur de notre santé, nous rendant tous super-centenaires, résistants à l’ensemble des maladies actuellement incurables et mortelles. Quel beau projet !
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Nous remercions Mme Karla Combes pour sa contribution à l’édition de cette revue. M.R. remercie le Dr Anita Krisko pour sa longue collaboration sur ces sujets et le Dr Adam de Graff pour les discussions stimulantes. Les travaux présentés ont été soutenus par la donation de Monsieur Jean-Noël Thorel et sa fondation, les contrats de recherche avec le groupe NAOS à Aix-en-Provence, France et Divide and Conquer, Cambridge, Royaume-Uni, ainsi que le Centre d’Excellence pour la Science et la Technologie (STIM), Split, Croatie.
Loi mathématique de distribution exponentielle de la mortalité.
La carbonylation correspond à la fixation de dérivés carbonylés (reactive carbonyl compounds, RCC) – des molécules contenant un groupement aldéhyde ou cétone – sur les protéines.
Références
- Krisko A, Radman M. Protein damage, ageing and age-related diseases. Open Biol 2019; 9 : 180249. [CrossRef] [PubMed] [Google Scholar]
- Radman M. Cellular parabiosis and the latency of age-related diseases. Open Biol 2019; 9. [Google Scholar]
- Krisko A, Radman M. Phenotypic and genetic consequences of protein damage. PLoS Genet 2013 ; 9. [Google Scholar]
- Kirkwood TB. Understanding the odd science of aging. Cell 2005 ; 120 : 437–447. [CrossRef] [PubMed] [Google Scholar]
- Krisko A, Radman M. Protein damage and death by radiation in Escherichia coli and Deinococcus radiodurans. Proc Natl Acad Sci USA 2010 ; 107 : 14373–14377. [CrossRef] [Google Scholar]
- Krisko A, Leroy M, Radman M, et al. Extreme anti-oxidant protection against ionizing radiation in bdelloid rotifers. Proc Natl Acad Sci USA 2012 ; 109 : 2354–2357. [CrossRef] [Google Scholar]
- Sulston JE, Brenner S. The DNA of Caenorhabditis elegans. Genetics 1974; 77 : 95-LP-104. [Google Scholar]
- Hengartner MO. Robert Horvitz H. Programmed cell death in Caenorhabditis elegans. Curr Opin Genet Dev 1994 ; 4 : 581–586. [CrossRef] [PubMed] [Google Scholar]
- Metzstein MM, Stanfield GM, Horvitz HR. Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet 1998 ; 14 : 410–416. [CrossRef] [PubMed] [Google Scholar]
- Krisko A, Radman M. Biology of extreme radiation resistance: the way of Deinococcus radiodurans. Cold Spring Harb Perspect Biol 2013 ; 5 : a012765–a012765. [CrossRef] [Google Scholar]
- López-otín C, Blasco MA, Partridge L, et al. The Hallmarks of aging longevity. Cell 2013 ; 153 : 1194–1217. [CrossRef] [PubMed] [Google Scholar]
- Speakman JR. Body size, energy metabolism and lifespan. J Exp Biol 2005; 208 : 1717-LP-30. [CrossRef] [Google Scholar]
- Karras GI, Yi S, Sahni N, et al. HSP90 shapes the consequences of human genetic variation. Cell 2017 ; 168 : 856–66 e12. [CrossRef] [Google Scholar]
- Bert P.. Expériences et considérations sur la greffe animale. J Anat Physiol 1864 ; 1 : 69–87. [Google Scholar]
- Nawaz M, Fatima F. Extracellular vesicles, tunneling nanotubes, and cellular interplay: synergies and missing links. Front Mol Biosci 2017 ; 4 : 1–12. [CrossRef] [PubMed] [Google Scholar]
- Gerdes HH, Bukoreshtliev N V, Barroso JFV. Tunneling nanotubes: a new route for the exchange of components between animal cells. FEBS Lett 2007 ; 581 : 2194–2201. [CrossRef] [PubMed] [Google Scholar]
- Vignais ML, Caicedo A, Brondello JM, et al. Cell connections by tunneling nanotubes: effects of mitochondrial trafficking on target cell metabolism, homeostasis, and response to therapy. Stem Cells Int 2017; 2017. [Google Scholar]
- Spees JL, Olson SD, Whitney MJ, et al. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA 2006 ; 103 : 1283–1288. [CrossRef] [Google Scholar]
- Orgel LE. The maintenance of the accuracy of protein synthesis and its relevance to ageing. Proc Natl Acad Sci USA 1963 ; 49 : 517–521. [CrossRef] [Google Scholar]
- Orgel LE. The maintenance of the accuracy of protein synthesis and its relevance to ageing: a correction. Proc Natl Acad Sci USA 1970 ; 67 : 1476. [CrossRef] [Google Scholar]
- Oliver CN, Ahn BW, Moerman EJ, et al. Age-related changes in oxidized proteins. J Biol Chem 1987 ; 262 : 5488–5491. [CrossRef] [PubMed] [Google Scholar]
- Stadtman ER. Protein oxidation and aging. Free Radic Res 2006 ; 40 : 1250–1258. [CrossRef] [PubMed] [Google Scholar]
- De Graff AMR, Hazoglou MJ, Dill KA.. Highly charged proteins: the Achilles’ heel of aging proteomes. Structure 2016 ; 24 : 329–336. [CrossRef] [PubMed] [Google Scholar]
- Castro JP, Ott C, Jung T, et al. Carbonylation of the cytoskeletal protein actin leads to aggregate formation. Free Radic Biol Med 2012 ; 53 : 91625. [CrossRef] [Google Scholar]
- Tanase M, Urbanska AM, Zolla V, et al. Role of carbonyl modifications on aging-associated protein aggregation. Sci Rep 2016 ; 6 : 1–14. [Google Scholar]
- Rahim A, Saha P, Jha KK, et al. Reciprocal carbonyl-carbonyl interactions in small molecules and proteins. Nat Commun 2017 ; 8 : 1–12. [CrossRef] [Google Scholar]
- Karri S, Singh S, Paripati AK, et al. Adaptation of Mge1 to oxidative stress by local unfolding and altered Interaction with mitochondrial Hsp70 and Mxr2. Mitochondrion 2019 ; 46 : 140–148. [CrossRef] [PubMed] [Google Scholar]
- Korovila I, Hugo M, Castro JP, et al. Proteostasis, oxidative stress and aging. Redox Biol 2017 ; 13 : 550–567. [CrossRef] [PubMed] [Google Scholar]
- Xu J, Reumers J, Couceiro JR, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol 2011 ; 7 : 285–295. [CrossRef] [Google Scholar]
- Sengupta U, Nilson AN, Kayed R. The role of amyloid-beta oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016 ; 6 : 42–49. [CrossRef] [PubMed] [Google Scholar]
- Ludtmann MHR, Angelova PR, Horrocks MH, et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat Commun 2018; 9. [Google Scholar]
Liste des figures
|
Figure 1. Interdépendance entre le protéome et le génome. L’ensemble des protéines (protéome) porte la fonction et assure le maintien de la vie. Dans le même temps, celles-ci assurent le maintien de l’ADN et la régulation de l’ensemble des gènes (génome) porteurs de l’information biologique. Le génome, quant à lui, assure la perpétuité et la pérennité de la vie, en maintenant le protéome par l’intermédiaire du transcriptome. Les protéines effectuent la synthèse, la réparation et le renouvellement des autres biomolécules. |
| Dans le texte | |
|
Figure 2. Corrélation entre mortalité cellulaire et carbonylation des protéines. Cette corrélation est observée pour deux types de bactéries : Escherichia coli (Eco) utilisée comme modèle, et Deinococcus radiodurans (Dra) qui est extrêmement résistante aux rayonnements (UV et g), utilisés pour induire la carbonylation des protéines et la mort. Pour deux espèces animales, le rotifère bdelloïde Adineta vaga qui est radiorésistant, et le ver modèle Caenorhabditis elegans, la mortalité est celles des cellules germinales (stérilité) (figure adaptée de [10]). |
| Dans le texte | |
|
Figure 3. Mauvais repliement et oxydation des protéines à l’origine des maladies liées au vieillissement. La protéine mal repliée ou mutante est plus sensible aux attaques par les espèces réactives de l’oxygène (ERO) et sera donc oxydée. Deux processus concurrents vont entrer en jeu : soit cette modification est réversible, et la protéine sera repliée dans sa conformation native par l’action des protéines chaperonnes, maintenant ainsi sa fonction (flèches vertes) ; soit la modification (carbonylation) fixe sa mauvaise conformation, qui n’est alors plus sujette à l’action des protéines chaperonnes (flèches rouges). La protéine oxydée et mal repliée peut alors suivre différentes voies : 1) soit elle est directement dégradée par les protéases ; 2) soit elle évite le système de dégradation de la cellule et reste sous forme de monomère dysfonctionnel, conduisant à la maladie (le cancer par exemple) ; 3) soit elle forme des structures multimériques (oligomères et agrégats) toxiques pour la cellule, à l’origine, entre autres, de l’apparition de maladies neurodégénératives. La présence d’agrégats immunogéniques induit une réponse du système immunitaire qui enclenche l’apparition et le développement des maladies liées au vieillissement. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.