")
")
| Issue |
Med Sci (Paris)
Volume 36, Number 6-7, Juin–Juillet 2020
Organoïdes
|
|
|---|---|---|
| Page(s) | 626 - 632 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2020098 | |
| Published online | 02 July 2020 | |
Les organoïdes de rétine
Un nouvel outil pour comprendre et traiter les maladies rétiniennes
Retinal organoids as a new tool for understanding and treating retinal diseases
Institut de la Vision, Sorbonne Université, Inserm, CNRS, 17 rue Moreau, F-75012 Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Les organoïdes de rétine dérivés de cellules souches pluripotentes représentent une avancée importante pour l’étude du développement de la rétine et offrent de nouvelles possibilités pour l’étude des maladies associées difficilement modélisables chez l’animal. La compréhension des étapes clefs du développement de la rétine chez les vertébrés a conduit à la mise au point de protocoles permettant d’obtenir, à partir de cellules souches pluripotentes, des structures tridimensionnelles auto-organisées contenant l’ensemble des types cellulaires de la rétine. Outre les applications en recherche fondamentale, ces organes miniatures ouvrent des perspectives encourageantes dans le domaine de la thérapie cellulaire ou le criblage de molécules thérapeutiques
Abstract
Generation of retinal organoids from pluripotent stem cells represents an important advance in the study of retinal development and offer new perspectives for the study of retinal diseases missing suitable animal models. Understanding the key stages of retinal development in vertebrates enabled to design protocols to generate self-organized three-dimensional structures derived from pluripotent stem cells and containing all retinal cell types. In addition to their application in basic research, such as the characterization of molecular and cellular mechanisms in retinal pathophysiology, these miniature organs also open up encouraging prospects in the field of cell therapy or the screening of therapeutic molecules, although some obstacles remain to be overcome.
© 2020 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Vignette (Photo © Olivier Goureau).
La possibilité de reprogrammer les cellules somatiques en cellules souches induites à la pluripotence (cellules iPS) et d’orienter ensuite ces cellules vers un destin cellulaire spécifique a ouvert un nouveau champ de recherches pour l’étude du développement anténatal humain, et celle des maladies humaines [1]. Les progrès concernant notre compréhension des programmes de développement, notamment grâce à la disponibilité de modèles animaux, et la maîtrise des protocoles in vitro de différenciation contrôlée des cellules souches pluripotentes humaines, permettent depuis quelques années de produire des structures tridimensionnelles présentant une organisation cellulaire et des propriétés proches de celle des organes. La formation de ces structures complexes, appelées organoïdes, repose sur la remarquable capacité des cellules souches pluripotentes à s’auto-organiser spatialement au cours de leur différenciation [2, 3] (→).
(→) Voir la Synthèse de J.L. Galzi et al., m/s n° 5, mai 2019, page 467
Si la fonctionnalité des organoïdes rétiniens obtenus à partir des protocoles actuels reste limitée en raison d’une relative immaturité, la rétinogenèse est en revanche assez fidèlement reproduite [4, 5]. Ces nouveaux modèles sont donc très intéressants pour l’étude du développement de la rétine dans un contexte humain et ouvrent de nouveaux champs d’investigation pour comprendre des maladies de la rétine difficilement modélisables chez l’animal, ainsi que des perspectives encourageantes dans le domaine de la thérapie cellulaire ou le criblage de molécules thérapeutiques.
Origine de l’œil et de la rétine
Les connaissances concernant le développement oculaire sont indispensables pour l’établissement de protocoles permettant de guider la différenciation des cellules souches pluripotentes humaines en cellules rétiniennes en conduisant à la formation d’organoïdes de rétine.
Au cours de l’embryogenèse, la rétine neurale et l’épithélium pigmentaire rétinien (EPR) dérivent du neurectoderme, la cornée et le cristallin de l’ectoderme de surface, et la sclère du mésoderme. La formation de l’œil s’amorce lors de la gastrulation par une série d’événements structuraux régis par des signaux moléculaires spécifiques qui confèrent à un groupe de cellules du neuroépithélium médian et antérieur une identité oculaire [6]. La délimitation de ce territoire dépend de l’activation des voies de signalisation du facteur de croissance des fibroblastes (FGF), du facteur de croissance apparenté à l’insuline (IGF-1) et de la répression de deux autres voies de transduction : la voie commune au facteur de croissance transformant bêta (TGF-β) et aux protéines morphogénétiques osseuses (BMP) et celle impliquant la signalisation du ligand Wnt (Figure 1A). La mise en place de ce territoire est orchestrée par la régulation temporelle et spatiale de gènes codant des facteurs de transcription spécifiques appelés « facteurs de transcription du champ oculaire » [6, 7] : six protéines homeobox différentes (LHX2, OTX2, PAX6, RAX, SIX3 et SIX6)1, un récepteur nucléaire (NR2E1 [nuclear receptor subfamily 2 group E member 1], aussi appelé TLX, pour tailless) et une protéine T-box (TBX3 pour T-box transcription factor 3). Sous l’influence de différents signaux, cette région va se scinder en deux pour former deux zones symétriques latérales. Ce remodelage se traduit par une évagination du neuroépithélium conduisant à la formation des vésicules optiques qui formeront, par la suite, la rétine et le nerf optique. Une étape d’invagination de ce neuroépithélium donnera naissance à la cupule optique, constituée désormais de deux couches (Figure 1B). Ces couches externe et interne formeront respectivement l’EPR et la neurorétine (aussi appelée rétine neurale) sous l’influence de signaux exogènes provenant de tissus adjacents, tels que le FGF, des agonistes de la voie TGF-β/BMP et le morphogène Sonic hedgehog (Shh) (Figure 1C). À ce stade, la future neurorétine est constituée de progéniteurs rétiniens multipotents, capables de donner naissance à tous les types cellulaires rétiniens selon une séquence temporelle qui est similaire chez tous les vertébrés [8]. Les types cellulaires précoces, comme les cellules ganglionnaires, les cellules amacrines et les cellules horizontales2, apparaissent tôt au cours du développement, tandis que les photorécepteurs, les cellules bipolaires et les cellules gliales de Müller sont générés à un stade plus avancé (Figure 1D–E).
 |
Figure 1. Les étapes du développement de la rétine des vertébrés. Au stade très précoce (A), sous l’influence de plusieurs gradients de facteurs morphogéniques, un territoire antérieur et médian de la plaque neurale dorsale acquiert une « identité oculaire ». Après formation du tube neural, ce territoire va se scinder en deux (B) et s’évaginer pour former les vésicules optiques. L’invagination de ces vésicules aboutit ensuite à la formation d’une structure présentant deux feuillets (C) qui formeront la future neurorétine et l’épithélium pigmentaire. À ce stade, la future neurorétine est constituée de progéniteurs multipotents (D) qui vont, par vagues successives, donner naissance aux différents types cellulaires qui constituent la neurorétine adulte (E). |
Produire des cellules et des tissus rétiniens à partir de cellules souches pluripotentes humaines
Cellules souches pluripotentes humaines
Les cellules souches pluripotentes sont caractérisées par l’association unique de deux propriétés qui sont mises à profit dans les domaines de l’ingénierie tissulaire et de la thérapie cellulaire : leur capacité d’auto-renouvellement (amplification clonale théoriquement infinie) et leur caractère pluripotent (potentiel de différenciation en n’importe quels types cellulaires constitutifs de l’organisme adulte). Depuis l’isolement des cellules souches embryonnaires (ES) humaines à la fin des années 1990 [9], et la mise au point de la technique dite des cellules iPS, dans les années 2006-2007 [10], la maîtrise des procédés permettant de guider le destin de ces cellules a progressé de façon spectaculaire.
Mimer le développement de l’œil
En utilisant le potentiel de différenciation des cellules ES et iPS humaines, il est possible de reproduire in vitro, de manière simplifiée, les mécanismes impliqués dans le développement de la rétine et de conduire les cellules pluripotentes à adopter une identité neuro-ectodermique, puis celle du territoire oculaire et, finalement, d’orienter leur différenciation vers un lignage rétinien (vers l’EPR ou vers la neurorétine). Cela a été rendu possible par l’utilisation séquentielle de composés chimiques ou de facteurs protéiques permettant l’activation successive des voies de signalisation du FGF et de l’IGF-1 ainsi que l’inhibition des deux voies de signalisation TGF-b/BMP et Wnt [4, 5].
Vers des mini-rétines en trois dimensions : les organoïdes rétiniens
Les premiers organoïdes de rétine ont été obtenus au début des années 2010 dans le laboratoire dirigé par Yoshiki Sasai au Ryken Center for Developmental Biology à Kobe au Japon [11]. L’un des éléments clés qui a permis à ces chercheurs d’obtenir non pas de « simples » cellules rétiniennes, mais des structures tridimensionnelles auto-organisées, a été la culture en suspension d’agrégats de cellules ES de souris dans des matrices extracellulaires artificielles. Une fois le processus de différenciation entamé, ces amas forment des corps embryonnaires. Ces chercheurs ont alors observé, en périphérie de ces corps embryonnaires, l’émergence de structures de type vésicule optique, contenant des progéniteurs rétiniens multipotents. Ces structures neurorétiniennes, après avoir été séparées des agrégats, peuvent poursuivre leur développement, laissant apparaître l’ensemble des types cellulaires rétiniens avec une organisation et une orientation semblables à celles observées in vivo. Ces auteurs ont obtenu des résultats similaires à partir de cellules ES humaines en 2012 [12]. Les organoïdes rétiniens ainsi obtenus présentaient une organisation en trois couches cellulaires regroupant des photorécepteurs à côté de plusieurs autres types neuronaux rétiniens et de cellules gliales de Müller. La relativement faible efficacité associée au coût élevé de cette méthode, en raison notamment du manque de contrôle de certains paramètres expérimentaux, a conduit plusieurs groupes à réduire cette hétérogénéité cellulaire initiale [13, 14]. Une approche alternative a consisté à contourner la formation des corps embryonnaires, en maintenant les cellules pluripotentes en conditions d’adhérence. Notre équipe a ainsi montré que les cellules iPS humaines à confluence, et en l’absence du facteur de pluripotence FGF2, pouvaient, une fois le processus de différenciation engagé, produire elles-mêmes des inhibiteurs des voies de signalisation TGF-β/BMP et Wnt, une situation favorable à la différenciation neuronale, et notamment rétinienne [15] (→).
(→) Voir la Nouvelle de S. Reichman et al., m/s n° 10, octobre 2014, page 845
L’utilisation de milieux classiquement utilisés pour la culture des neurones favorise également l’émergence de structures auto-organisées de type vésicule optique. La mise en culture en suspension de ces structures permet, dans un second temps, la formation d’organoïdes rétiniens contenant l’ensemble des types cellulaires de la rétine [16, 17]. Comme dans de nombreux protocoles de différenciation en trois dimensions (3D), on peut distinguer trois étapes successives dans l’évolution structurale des organoïdes rétiniens (Figure 2). Les premiers organoïdes, âgés de 35-40 jours, sont constitués d’un neuro-épithélium contenant des progéniteurs rétiniens en prolifération. Aux stades intermédiaires (entre 60 et 100 jours), une séparation nette entre une couche nucléaire externe et une couche interne présomptives, correspondant à la différenciation des photorécepteurs et des populations de neurones de la rétine interne, est observée. Enfin, dans les organoïdes rétiniens d’âge tardif (après 150-175 jours), les photorécepteurs peuvent atteindre un stade avancé de développement, comme l’illustre la formation de segments internes et externes. On peut souligner le fait que cette cinétique est assez proche de celle observée au cours du développement fœtal humain (entre les semaines 23 et 30). Certaines limites persistent toutefois, notamment lorsqu’il s’agit de maintenir à long terme ces organoïdes. Une désorganisation progressive de la région interne des organoïdes aux stades tardifs est en effet observée, ainsi qu’une perte des cellules ganglionnaires, prévisible dans ce type de cultures en l’absence des cibles cérébrales naturelles de ces neurones.
 |
Figure 2. Les différents stades des organoïdes rétiniens générés à partir de cellules souches pluripotentes humaines. Diagramme montrant l’évolution structurale, la stratification et la composition cellulaire des organoïdes au cours du temps. |
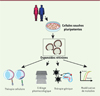 |
Figure 3. Les principales applications d’organoïdes rétiniens dérivés de cellules souches pluripotentes. L’utilisation d’organoïdes peut répondre à différentes problématiques en recherche fondamentale et en médecine réparatrice (thérapie cellulaire et génique). La génération de modèles associés aux maladies génétiques permet d’étudier le développement de ces maladies (modélisation) et leur réponse à différents traitements (criblage pharmacologique). |
Les progrès récents réalisés dans les approches de séquençage des ARN messagers à l’échelle d’une seule cellule ont permis de caractériser précisément les différentes populations rétiniennes au cours de la différenciation des organoïdes et de montrer la similarité entre la rétinogenèse in vitro (organoïdes) et in vivo (tissus fœtaux humains) [18, 19]. Plusieurs études ont montré la capacité des cellules ganglionnaires présentes dans des organoïdes relativement jeunes à émettre des potentiels d’action traduisant une certaine maturité fonctionnelle [20, 21]. D’autres études ont rapporté l’existence de réponse à la lumière par les photorécepteurs dans les organoïdes de plus de 25 semaines [14], ou une modification des niveaux de calcium au sein des photorécepteurs après application de nucléotides cycliques dérivés du GTP (GMPc), une caractéristique de la photo-transduction, indiquant dans les deux cas, une maturation fonctionnelle des photorécepteurs [17, 22].
Organoïdes de rétines comme sources de cellules pour la thérapie cellulaire
Outre leur intérêt pour étudier le développement humain, les organoïdes rétiniens représentent une source très intéressante de cellules d’intérêt thérapeutique. Dans une démarche de médecine dite régénérative, ou thérapie cellulaire, on peut envisager la production de cellules spécifiques qui pourraient être implantées chez des patients souffrant d’une perte de ces cellules. Des publications récentes décrivent la transplantation dans différents modèles animaux de photorécepteurs issus d’organoïdes, en suivant différentes stratégies [5, 22-25]. Certaines approches consistent à transplanter une population purifiée de photorécepteurs immatures, après une étape de sélection (tri cellulaire par cytométrie en flux ou magnétique) [22, 24] ; d’autres proposent la greffe d’un feuillet de rétine issu d’organoïdes rétiniens dans des modèles animaux où l’ensemble des photorécepteurs ont dégénéré [24, 25]. Toutefois, la capacité des photorécepteurs greffés à se reconnecter dans le tissu hôte est très réduite en raison de la présence, au sein de ces feuillets, de cellules rétiniennes autres que les photorécepteurs. Quelle que soit la stratégie employée, la capacité limitée des cellules greffées à se développer en photorécepteurs fonctionnels (sensibles à la lumière) connectés à la fois à la rétine interne et à l’EPR de l’hôte reste un obstacle majeur. De nouvelles voies de recherche, visant à combiner la thérapie cellulaire et l’optogénétique ou la bio-ingénierie (polymères structuraux biocompatibles ou échaffaudages [scaffold]), commencent à émerger. L’utilisation de scaffold a pour but de favoriser la maturation et la polarisation des photorécepteurs afin de transplanter des photorécepteurs avec des segments externes sensibles à la lumière [26]. L’approche optogénétique permet de rendre les cellules artificiellement sensibles à la lumière, grâce à l’expression d’opsines3 microbiennes spécifiques, même en l’absence de maturation fonctionnelle des photorécepteurs [27].
Organoïdes de rétines comme outils de modélisation pour la compréhension de maladies et pour le criblage de médicaments
Dans la mesure où les organoïdes peuvent être obtenus à partir de cellules iPS de patients atteints de maladies génétiques, ils peuvent être utilisés pour comprendre le processus pathologique conduisant à la maladie, et comme outil pour le développement de nouvelles thérapies pharmacologiques ou géniques.
Concernant les maladies de la rétine, la technique des cellules iPS est particulièrement intéressante dans le cas des maladies dégénératives héréditaires, telles que les rétinites pigmentaires. Bien que des modèles murins puissent être utilisés pour étudier certains aspects de ces maladies, ils ne présentent pas toujours les signes cliniques de la maladie ou ne reproduisent pas fidèlement le processus pathologique, et leur rétine ne possèdent pas toutes les caractéristiques d’une rétine humaine. Ainsi, les cellules iPS obtenues à partir de patients ont permis de produire des organoïdes présentant des phénotypes pathologiques, comme une perte prématurée des photorécepteurs comparable à celle observée chez les sujets atteints [28]. Ces organoïdes sont aujourd’hui utilisés pour comprendre les mécanismes moléculaires et cellulaires responsables de la mort des photorécepteurs menant à la cécité chez les patients.
L’accès à des biopsies de patients volontaires n’est pas toujours facile, notamment pour les maladies rares. La nouvelle technique d’édition du génome CRISPR/Cas9 peut permettre aujourd’hui de s’en affranchir. La palette d’outils liée à cette technique rend en effet possible la modification ciblée, in vitro, du génome pour reproduire au sein de cellules iPS « normales » des mutations connues pour être responsables de maladies [29], et ainsi de générer des organoïdes rétiniens porteurs de mutation(s) avec un phénotype pathologique. Il est aussi possible d’utiliser cette technique pour corriger le(s) mutation(s) et valider la réversion d’un phénotype au sein des organoïdes [29, 30].
Une autre application intéressante consiste à utiliser des organoïdes de rétine pour le criblage automatisé à haut débit de centaines de milliers de composés chimiques, afin d’identifier des molécules d’intérêt « thérapeutique », à une échelle qui ne pourrait pas être réalisée sur des modèles animaux. Toutefois, le défi de miniaturisation et d’automatisation reste important en raison de la complexité des cultures d’organoïdes (longues étapes de différenciation, traitements nécessaires pour l’imagerie, etc.).
Se rapprocher d’une vraie rétine en augmentant le degré de complexité des organoïdes rétiniens
Malgré les similitudes qui ont pu être obtenues entre les organoïdes rétiniens et une rétine adulte, les niveaux d’organisation et de fonctionnalité de ces deux entités sont loin d’être identiques ; ce qui est tout à fait compréhensible étant donné les différences marquées entre les environnements in vitro et in vivo. Des améliorations futures pourraient reposer sur l’enrichissement des systèmes de culture. Par exemple, les organoïdes rétiniens isolés ne forment pas une véritable cupule optique et ne présentent pas un EPR correctement localisé, en face des photorécepteurs. La coculture d’organoïdes en présence d’EPR pourrait améliorer la morphogenèse des segments externes des photorécepteurs [31] et, au final, leur fonction (réponse à la lumière), ou permettre de modéliser des maladies caractérisées par des défauts d’interaction entre ces deux tissus. Les organoïdes ne sont pas non plus reliés au cerveau antérieur, ou à d’autres structures non-rétiniennes importantes pour le développement rétinien. Le maintien de connexions rétine-cerveau par l’intermédiaire de cocultures (ou « assembloïdes ») d’organoïdes rétiniens et d’organoïdes cérébraux pourrait faciliter la survie des cellules ganglionnaires et ainsi permettre la formation d’une structure semblable au nerf optique.
Une des limites des protocoles développés pour la formation des organoïdes de rétine, comme pour ceux utilisés pour les organoïdes cérébraux, est qu’ils dirigent les cellules iPS dans un lignage neurectodermique, en inhibant la formation du mésoderme et de l’endoderme. Les organoïdes rétiniens ne présentent donc pas la combinaison complète de cellules dérivées des différentes couches germinales qui sont présentes in vivo dans une rétine, telles que les cellules microgliales et les cellules vasculaires. Cette absence de vascularisation des organoïdes rétiniens limite ainsi le soutien métabolique au centre des structures à des stades avancés (lorsque leur taille est supérieure à 250 µm) et pourrait expliquer la nécrose observée, au fil du temps, au cœur des organoïdes. Des approches de cocultures avec des cellules vasculaires, ou de bio-ingénierie tissulaire (échafaudages biologiques, bio-impression, etc.) pourraient permettre la formation de néo-vaisseaux dans les organoïdes. Une approche alternative peut être la greffe de ces organoïdes produits in vitro dans un animal hôte, qui permettra l’approvisionnement en nutriments dans un environnement adéquat, comme cela a été rapporté récemment pour des organoïdes cérébraux [32].
L’ensemble de ces considérations doit être pris en compte, notamment pour les approches de modélisation, qui, par définition, cherchent à reproduire le plus fidèlement possible la situation naturelle in vivo. À titre d’exemple, la distribution spatiale particulière des différents sous-types de photorécepteurs chez l’homme, qui se caractérise par un regroupement des cônes au sein de la partie centrale de la rétine, dans la région maculaire (ou fovéa), n’est actuellement pas observée dans les organoïdes rétiniens humains.
Conclusion
L’émergence de protocoles fondés sur la différenciation des cellules iPS en organoïdes de rétine qui regroupent, in vitro, les principaux évènements moléculaires et cellulaires de la rétinogenèse humaine a ouvert d’immenses possibilités de recherche sur le développement, la modélisation et l’étude de processus pathologiques ou la production de cellules pertinentes en clinique. Encore imparfaits à ce jour, ils sont néanmoins complémentaires des modèles animaux, qui présentent d’autres avantages mais ont leurs limites. La mise au point d’assembloïdes, en combinant plusieurs types cellulaires ou d’organoïdes (rétine, cerveau, EPR, vaisseaux, etc.) constitue une piste intéressante pour le développement de modèles 3D ex vivo encore plus fidèles à la réalité complexe in vivo.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
LIM homeobox 2, orthodenticle homeobox 2, paired box 6, retina and anterior neural fold homeobox, SIX homeobox. Protéines impliquées dans le développement.
Les cellules horizontales partagent avec les cellules amacrines une caractéristique particulière : l’absence de prolongement similaire à un axone. Ces cellules ne possèdent que des dendrites dont certaines peuvent être présynaptiques, et donc jouer le rôle d’axone.
Protéines capables de réagir à l’énergie lumineuse et provoquant directement ou indirectement la modification de courants traversant la cellule.
Références
- Karagiannis P, Takahashi K, Saito M, et al. Induced pluripotent stem cells and their use in human models of disease and development. Physiol Rev 2019 ; 99 : 79–114. [Google Scholar]
- Rossi G, Manfrin A, Lutolf MP. Progress and potential in organoid research. Nat Rev Genet 2018 ; 19 : 671–687. [CrossRef] [PubMed] [Google Scholar]
- Galzi J, Jouault T, Amédée J. Les organoïdes : des mini-organes au service de la biomédecine. Med Sci (paris) 2019 ; 35 : 467–469. [Google Scholar]
- Llonch S, Carido M, Ader M. Organoid technology for retinal repair. Dev Biol 2018 ; 433 : 132–143. [CrossRef] [PubMed] [Google Scholar]
- Gagliardi G, M’Barek K. Ben, Goureau O. Photoreceptor cell replacement in macular degeneration and retinitis pigmentosa: a pluripotent stem cell-based approach. Prog. Retin. Eye Res 2019 ; 71 : 1–25. [Google Scholar]
- Graw J. Eye development. Curr Top Dev Biol 2010 ; 90 : 343–386. [CrossRef] [Google Scholar]
- Zuber ME, Cagan RL, Ross L, Reh TA. Eye Field specification in Xenopus laevis. Current topics in developmental biology 2010 ; New York Elsevier Inc 29–60. [Google Scholar]
- Cepko C.. Intrinsically different retinal progenitor cells produce specific types of progeny. Nat Rev Neurosci 2014 ; 15 : 615–627. [CrossRef] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998 ; 282 : 1145–1147. [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007 ; 131 : 861–872. [CrossRef] [PubMed] [Google Scholar]
- Eiraku M, Takata N, Ishibashi H, et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011 ; 472 : 51–56. [CrossRef] [PubMed] [Google Scholar]
- Nakano T, Ando S, Takata N, et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012 ; 10 : 771–785. [Google Scholar]
- Meyer JS, Howden SE, Wallace KA, et al. Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells 2011 ; 29 : 1206–1218. [CrossRef] [PubMed] [Google Scholar]
- Zhong X, Gutierrez C, Xue T, et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun 2014 ; 5 : 4047. [CrossRef] [PubMed] [Google Scholar]
- Reichman S, Sahel JA, Goureau O. Production de rétines in vitro à partir de cellules pluripotentes humaines. Med Sci (Paris) 2014 ; 30 : 845–848. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Reichman S, Terray A, Slembrouck A, et al. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc Natl Acad Sci USA 2014 ; 111 : 8518–8523. [CrossRef] [Google Scholar]
- Reichman S, Slembrouck A, Gagliardi G, et al. Generation of storable retinal organoids and retinal pigmented epithelium from adherent human iPS cells in xeno-free and feeder-free conditions. Stem Cells 2017 ; 35 : 1176–1188. [CrossRef] [PubMed] [Google Scholar]
- Mao X, An Q, Xi H, et al. Single-cell RNA sequencing of hESC-derived 3D retinal organoids reveals novel genes regulating RPC commitment in early human retinogenesis. Stem Cell Rep 2019 ; 13 : 747–760. [CrossRef] [Google Scholar]
- Kim S, Lowe A, Dharmat R, et al. Generation, transcriptome profiling, and functional validation of cone-rich human retinal organoids. Proc Natl Acad Sci USA 2019 ; 116 : 10824–10133. [CrossRef] [Google Scholar]
- Rabesandratana O, Goureau O, Orieux G. Pluripotent stem cell-based approaches to explore and treat optic neuropathies. Front Neurosci 2018 ; 12 : 651. [Google Scholar]
- VanderWall KB, Vij R, Ohlemacher SK, et al. Astrocytes regulate the development and maturation of retinal ganglion cells derived from human pluripotent stem cells. Stem Cell Rep 2019 ; 12 : 201–212. [CrossRef] [Google Scholar]
- Gagliardi G, Ben M’Barek K, Chaffiol A, et al. Characterization and transplantation of cd73-positive photoreceptors isolated from human iPSc-derived retinal organoids. Stem Cell Rep 2018 ; 11 : 665–680. [CrossRef] [Google Scholar]
- Gonzalez-Cordero A, Kruczek K, Naeem A, et al. Recapitulation of human retinal development from human pluripotent stem cells generates transplantable populations of cone photoreceptors. Stem Cell Rep 2017 ; 9 : 820–837. [CrossRef] [Google Scholar]
- Gasparini SJ, Llonch S, Borsch O, et al. Transplantation of photoreceptors into the degenerative retina: current state and future perspectives. Prog Retin Eye Res 2019 ; 69 : 1–37. [CrossRef] [PubMed] [Google Scholar]
- Shirai H, Mandai M, Matsushita K, et al. Transplantation of human embryonic stem cell-derived retinal tissue in two primate models of retinal degeneration. Proc Natl Acad Sci USA 2016 ; 113 : E81–E90. [CrossRef] [Google Scholar]
- Jung YH, Phillips MJ, Lee J, et al. 3D microstructured scaffolds to support photoreceptor polarization and maturation. Adv Mater 2018 ; 30 : 1803550. [Google Scholar]
- Garita-Hernandez M, Lampicˇ M, Chaffiol A, et al. Restoration of visual function by transplantation of optogenetically engineered photoreceptors. Nat Commun 2019 ; 10 : 4524. [CrossRef] [PubMed] [Google Scholar]
- Artero Castro A, Lukovic D, Jendelova P, et al. Concise review: human induced pluripotent stem cell models of retinitis pigmentosa. Stem Cells 2018; 36 : 474–81. [CrossRef] [PubMed] [Google Scholar]
- Burnight ER, Giacalone JC, Cooke JA, et al. CRISPR-Cas9 genome engineering: treating inherited retinal degeneration. Prog Retin Eye Res 2018 ; 65 : 28–49. [CrossRef] [PubMed] [Google Scholar]
- Deng WL, Gao ML, Lei XL, et al. Gene correction reverses ciliopathy and photoreceptor loss in ipsc-derived retinal organoids from retinitis pigmentosa patients. Stem Cell Rep 2018 ; 10 : 1267–1281. [CrossRef] [Google Scholar]
- Achberger K, Probst C, Haderspeck J, et al. Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human retina-on-a-chip platform. Elife 2019; 8 [Google Scholar]
- Mansour AA, Gonçalves JT, Bloyd CW, et al. An in vivo model of functional and vascularized human brain organoids. Nat Biotechnol 2018 ; 36 : 432–441. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Les étapes du développement de la rétine des vertébrés. Au stade très précoce (A), sous l’influence de plusieurs gradients de facteurs morphogéniques, un territoire antérieur et médian de la plaque neurale dorsale acquiert une « identité oculaire ». Après formation du tube neural, ce territoire va se scinder en deux (B) et s’évaginer pour former les vésicules optiques. L’invagination de ces vésicules aboutit ensuite à la formation d’une structure présentant deux feuillets (C) qui formeront la future neurorétine et l’épithélium pigmentaire. À ce stade, la future neurorétine est constituée de progéniteurs multipotents (D) qui vont, par vagues successives, donner naissance aux différents types cellulaires qui constituent la neurorétine adulte (E). |
| Dans le texte | |
|
Figure 2. Les différents stades des organoïdes rétiniens générés à partir de cellules souches pluripotentes humaines. Diagramme montrant l’évolution structurale, la stratification et la composition cellulaire des organoïdes au cours du temps. |
| Dans le texte | |
|
Figure 3. Les principales applications d’organoïdes rétiniens dérivés de cellules souches pluripotentes. L’utilisation d’organoïdes peut répondre à différentes problématiques en recherche fondamentale et en médecine réparatrice (thérapie cellulaire et génique). La génération de modèles associés aux maladies génétiques permet d’étudier le développement de ces maladies (modélisation) et leur réponse à différents traitements (criblage pharmacologique). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.