")
")
| Issue |
Med Sci (Paris)
Volume 35, Number 11, Novembre 2019
|
|
|---|---|---|
| Page(s) | 836 - 838 | |
| Section | Le Magazine | |
| DOI | https://doi.org/10.1051/medsci/2019163 | |
| Published online | 17 December 2019 | |
Le cil primaire au cœur de la pathogénie du prolapsus de la valve mitrale
The primary cilia at the heart of mitral valve prolapse pathogeny
Inserm U1251, université Aix-Marseille, MMG, 27 boulevard Jean Moulin, 13885 Marseille, France
* This email address is being protected from spambots. You need JavaScript enabled to view it.
Les malformations congénitales des valves du cœur ont une prévalence estimée à environ 5 % des nouveau-nés, et sont présentes dans 20 % à 30 % des cas de malformation cardio-vasculaire congénitale [1]. La valve mitrale est souvent affectée. Le prolapsus de la valve mitrale plus modéré et sans symptôme est par ailleurs fréquent dans la population générale, affectant 1 individu sur 40, tous âges confondus [2]. Le prolapsus de la valve mitrale congénital non syndromique résulte d’une anomalie du développement embryonnaire de cette valve. Ses conséquences sur la fonction cardiaque ne s’observent cependant, pour la plupart des sujets atteints, qu’à l’âge adulte. Des études génétiques ont identifié, chez les individus atteints, des mutations dans les gènes codant la filamine A (FLNA) [3], la LIM and cysteine-rich domains 1 (LMCD1), la tensine 1 (TNS1) [4], Dachsous (DCHS1) [5], et la zinc-finger protein (DZIP1) [6], mais ces mutations ne rendent compte qu’à peine de 2 % des cas et leurs effets sur le développement de la valve mitrale restent à déterminer. La malformation conduisant au prolapsus de la valve mitrale résulte d’une sécrétion excessive de protéines de la matrice extracellulaire par les cellules interstitielles dans les feuillets valvulaires, ce qui entraîne une dégénérescence myxomateuse ou fibro-élastique de la valve. La fuite de sang vers l’oreillette gauche lors de la contraction ventriculaire (régurgitation mitrale) [7] altère progressivement la fonction du ventricule gauche, nécessitant alors une intervention chirurgicale pour réparer ou changer la valve.
Nous nous sommes intéressés au lien intrigant entre la mutation du gène DCHS1, qui code Dachsous, une protéine impliquée dans la polarité planaire des épithéliums, et le prolapsus de la valve mitrale. En effet, dans le cadre du réseau transatlantique d’excellence MITRAL financé par la Fondation Leducq, nos collaborateurs français et américains avaient identifié, en France et aux États-Unis, deux familles comportant plusieurs individus porteurs, à l’état hétérozygote, d’une mutation faux-sens dans le gène DCHS1 (c.6988C>T ; p.Arg2330Cys ou c.7538G>A ; p.Arg2513His) et atteints d’un prolapsus de la valve mitrale [5].
Différenciation de cellules souches pluripotentes induites en cellules valvulaires
Nous avons produit des cellules souches pluripotentes ou cellules iPS (induced pluripotent stem cells) à partir de cellules valvulaires d’un patient porteur de la mutation DCHS1:c.6988C>T prélevées lors d’une intervention chirurgicale sur sa valve mitrale. Des cellules iPS non mutées et les cellules iPS porteuses de la mutation ont été différenciées en cellules endocardiques, puis en cellules valvulaires, par un protocole mimant le scénario du développement normal des valves [8] pendant l’embryogenèse [9] (Figure 1). Les cellules iPS non mutées ont d’abord été différenciées en cellules du mésoderme par l’action de la protéine BMP2 (bone morphogenetic protein 2) et de la protéine Wnt3a (Wingless integration site 3a), puis en cellules endocardiques par l’action du VEGF (vascular endothelial growth factor) et du FGF8 (fibroblast growth factor 8). Afin de caractériser le phénotype de ces cellules, nous avons comparé leur transcriptome et celui des cellules endocardiques du canal atrio-ventriculaire de souris au stade embryonnaire E9.5, au moment de la transition épithélio-mésenchymateuse d’une fraction des cellules endocardiques qui est à l’origine des coussins puis des feuillets valvulaires de la valve mitrale [8]. Cette comparaison a révélé de nombreux transcrits communs aux deux préparations cellulaires. Nous avons confirmé ce résultat en déterminant la séquence nucléotidique des ARN messagers issus de cellules uniques. Cette analyse a révélé l’hétérogénéité des cellules endocardiques obtenues par différenciation des cellules iPS, incluant des cellules hémogéniques1 (une possibilité connue de différenciation des cellules endocardiques [10]), mais aussi des cellules endothéliales ou en cours de transition épithélio-mésenchymateuse, c’est-à-dire les cellules à l’origine des valves. Cette analyse a également montré la fiabilité du protocole de différenciation des cellules iPS in vitro (Figure 1). Afin de mimer la situation physiologique, les cellules endocardiques obtenues à partir des cellules iPS non mutées ont ensuite été cultivées en présence de BMP2 (qui est naturellement sécrétée par les cellules du myocarde in vivo) pour déclencher la transition épithélio-mésenchymateuse convertissant ces cellules endocardiques en cellules valvulaires interstitielles. Le séquençage des ARN messagers issus de cellules uniques a montré la diversité des cellules valvulaires interstitielles obtenues, et notamment un début de différenciation vers les types cellulaires attendus dans les différentes couches histologiques de la valve mitrale : fibrosa (riche en protéoglycanes), spongiosa (riche en fibres de collagène), et lamina atrialis ou lamina ventricularis, qui sont les deux couches les plus superficielles situées respectivement du côté atrial et du côté ventriculaire de la valve (riches en fibres d’élastine et de collagène) [11]. L’analyse bioinformatique de ces transcriptomes a montré que les cellules interstitielles se regroupaient en fonction de leur identité cellulaire et de leur localisation future dans la valve.
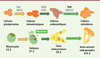 |
Figure 1 La différenciation de cellules pluripotentes humaines in vitro mime la valvulogenèse embryonnaire chez la souris. TEM : transition épithélio-mésenchymateuse ; V : ventricule primaire ; O : oreillette ; CAV : canal atrio-ventriculaire ; TE : tronc efférent. |
La différenciation des cellules iPS porteuses de la mutation de DCHS1 en cellules endocardiques par le même protocole a montré tout d’abord que les cellules obtenues avaient conservé leur capacité de transition épithélio-mésenchymateuse en présence de BMP2. Des analyses de protéines de la matrice extracellulaire (collagène, hyaluronane et périostine) par immunofluorescence ont montré une sécrétion accrue de ces protéines par ces cellules. De plus, l’analyse du transcriptome de cellules uniques a montré une perte d’identité des cellules valvulaires interstitielles mutées. Toutes exprimaient tous les gènes codant les protéines de la matrice extracellulaire, et il n’était plus possible de regrouper ces cellules en sous-types correspondant aux différentes couches histologiques. Enfin, l’analyse du cil primaire des cellules interstitielles mutées a révélé un nombre plus faible de cellules ciliées et la présence de cils plus courts, comme cela avait été montré précédemment chez les souris hétérozygotes invalidées pour le gène Dchs1 [5].
Le cil primaire au cœur de la pathogénie du prolapsus de la valve mitrale
Le cil primaire transmet à la cellule des signaux extérieurs par l’intermédiaire de différents récepteurs membranaires et voies de transduction, dont la voie de Sonic hedgehog (SHH) et son récepteur membranaire Patched 1 [12], connue pour activer l’expression de gènes codant des protéines de la matrice extracellulaire [13]. SHH, par sa liaison à Patched 1 à la surface du cil, induit la translocation de ce récepteur à la surface apicale de la cellule, ce qui déclenche la signalisation intracellulaire. Nous avons fait l’hypothèse que les cellules valvulaires interstitielles porteuses de la mutation de DCHS1 dépourvues de cil primaire ou possédant un cil anormalement court étaient incapables de transduire correctement le signal SHH. Une analyse de la localisation de Patched 1 par immunofluorescence a en effet montré que ce récepteur, normalement situé sur le cil primaire des cellules non mutées en l’absence du ligand, était anormalement présent à la surface apicale des cellules porteuses de la mutation, où son activation constitutive devait stimuler la production de protéines de la matrice extracellulaire. Nous avons alors testé l’efficacité d’un inhibiteur pharmacologique de la voie de SHH, la cyclopamine, sur des cellules mutées, ce qui a permis de rétablir un phénotype cellulaire normal. Inversement, une activation de la voie SHH dans les cellules non mutées par l’ajout d’une forte dose du ligand entraîne un phénotype cellulaire anormal (Figure 2).
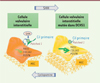 |
Figure 2 Mécanisme d’activation constitutive de la voie de signalisation de sonic hedgehog (SHH) et de la sécrétion excessive de protéines de la matrice extracellulaire dans les cellules valvulaires mutées. Dans la cellule valvulaire interstitielle normale (à gauche), le récepteur Patched 1, après liaison de son ligand SHH, se déplace du cil primaire vers la surface apicale pour activer la voie de signalisation intracellulaire. SHH active l’expression de gènes codant des protéines de la matrice extracellulaire. Dans la cellule porteuse de la mutation de DCHS1 (à droite), Patched 1 est anormalement localisé à la surface apicale même en l’absence de SHH, et la voie de signalisation est constamment activée. La cyclopamine, qui inhibe la voie de signalisation de SHH, corrige le phénotype cellulaire anormal. Inversement, une suractivation de la voie SHH dans des cellules non mutées leur confère un phénotype pathologique. MEC : matrice extracellulaire ; HAS1 : gène codant la hyaluronane synthase 1 ; COL1A1 : gène codant la chaîne alpha-1 du collagène 1 ; POSTN : gène codant la périostine. |
Perspectives
La voie de signalisation par SHH peut être manipulée pharmacologiquement. Elle joue un rôle majeur au cours du développement embryonnaire, et est moins active dans l’organisme adulte (en dehors de certaines maladies telles que le cancer ou la fibrose tissulaire). Une inhibition de la voie SHH pourrait constituer une nouvelle opportunité thérapeutique pour le prolapsus de la valve mitrale chez les individus porteurs de mutations des gènes DCHS1 ou DZIP1 [6]. On ignore encore si cette voie de signalisation est impliquée dans d’autres formes de prolapsus valvulaire. ◊
Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Cellule hémogénique : cellule capable de différenciation en cellule hématopoïétique.
Références
- Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res 2009 ; 105 : 408–21. [Google Scholar]
- Levine RA, Hagège AA, Judge DP, et al. Mitral valve disease : morphology and mechanisms. Nat Rev Cardiol 2015 ; 12 : 689–710. [Google Scholar]
- Kyndt F, Gueffet JP, Probst V, et al. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation 2007 ; 115 : 40–9. [CrossRef] [PubMed] [Google Scholar]
- Dina C, Bouatia-Naji N, Tucker N, et al. Genetic association analyses highlight biological pathways underlying mitral valve prolapse. Nat Genet 2015 ; 47 : 1206–11. [Google Scholar]
- Durst R, Sauls K, Peal DS, et al. Mutations in DCHS1 cause mitral valve prolapse. Nature 2015 ; 525 : 109–13. [Google Scholar]
- Toomer KA, Yu M, Fulmer D, et al. Primary cilia defects causing mitral valve prolapse. Sci Transl Med 2019 ; 11 : 493. [Google Scholar]
- Delling FN, Vasan RS. Epidemiology and pathophysiology of mitral valve prolapse: new insights into disease progression, genetics, and molecular basis. Circulation 2014 ; 129 : 2158–70. [CrossRef] [PubMed] [Google Scholar]
- Puceat M. Embryological origin of the endocardium and derived valve progenitor cells: from developmental biology to stem cell-based valve repair. Biochim Biophys Acta 2013 ; 1833 : 917–22. [CrossRef] [PubMed] [Google Scholar]
- Neri T, Hiriart E, van Vliet PP, et al. Human pre-valvular endocardial cells derived from pluripotent stem cells recapitulate cardiac pathophysiological valvulogenesis. Nat Commun 2019 ; 10 : 1929. [CrossRef] [PubMed] [Google Scholar]
- Van Handel B, Montel-Hagen A, Sasidharan R, et al. Scl represses cardiomyogenesis in prospective hemogenic endothelium and endocardium. Cell 2012 ; 150 : 590–605. [CrossRef] [PubMed] [Google Scholar]
- Schlotter F, Halu A, Goto S, et al. Spatiotemporal multi-omics mapping generates a molecular atlas of the aortic valve and reveals networks driving disease. Circulation. 2018 ; 138 : 377–93. [CrossRef] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP, Patched1 regulates hedgehog signaling at the primary cilium. Science 2007 ; 317 : 372–6. [Google Scholar]
- Liu J, Li Q, Kuehn MR, et al. Sonic hedgehog signaling directly targets hyaluronic acid synthase 2, an essential regulator of phalangeal joint patterning. Dev Biol 2013 ; 375 : 160–71. [CrossRef] [PubMed] [Google Scholar]
© 2019 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (http://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (http://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l'utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Liste des figures
|
Figure 1 La différenciation de cellules pluripotentes humaines in vitro mime la valvulogenèse embryonnaire chez la souris. TEM : transition épithélio-mésenchymateuse ; V : ventricule primaire ; O : oreillette ; CAV : canal atrio-ventriculaire ; TE : tronc efférent. |
| Dans le texte | |
|
Figure 2 Mécanisme d’activation constitutive de la voie de signalisation de sonic hedgehog (SHH) et de la sécrétion excessive de protéines de la matrice extracellulaire dans les cellules valvulaires mutées. Dans la cellule valvulaire interstitielle normale (à gauche), le récepteur Patched 1, après liaison de son ligand SHH, se déplace du cil primaire vers la surface apicale pour activer la voie de signalisation intracellulaire. SHH active l’expression de gènes codant des protéines de la matrice extracellulaire. Dans la cellule porteuse de la mutation de DCHS1 (à droite), Patched 1 est anormalement localisé à la surface apicale même en l’absence de SHH, et la voie de signalisation est constamment activée. La cyclopamine, qui inhibe la voie de signalisation de SHH, corrige le phénotype cellulaire anormal. Inversement, une suractivation de la voie SHH dans des cellules non mutées leur confère un phénotype pathologique. MEC : matrice extracellulaire ; HAS1 : gène codant la hyaluronane synthase 1 ; COL1A1 : gène codant la chaîne alpha-1 du collagène 1 ; POSTN : gène codant la périostine. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.