")
")
| Issue |
Med Sci (Paris)
Volume 33, Number 6-7, Juin-Juillet 2017
|
|
|---|---|---|
| Page(s) | 674 - 676 | |
| Section | Forum | |
| DOI | https://doi.org/10.1051/medsci/20173306028 | |
| Published online | 19 July 2017 | |
Variants fréquents et rares, caractères multigéniques et héritabilité perdue
Chroniques génomiques
Common and rare variants, polygenic traits and missing heritability
UMR 7268 ADÉS, Aix-Marseille, Université/EFS/CNRS,Espace éthique méditerranéen, hôpital d’adultes la Timone, 264, rue Saint-Pierre, 13385 Marseille Cedex 05, France ;
CoReBio PACA, case 901, parc scientifique de Luminy, 13288 Marseille Cedex 09, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
**
This email address is being protected from spambots. You need JavaScript enabled to view it.
Abstract
Recently, a systematic (but limited) search for rare variants implicated in adult height, a highly polygenic trait, has uncovered a number of new variants for which the effect size is inversely correlated with the minor allele frequency. This opens interesting perspectives on the genetic architecture of complex traits and on the vexing problem of “missing heritability”.
© 2017 médecine/sciences – Inserm

Un article récemment paru dans Nature [1] revient sur les déterminants génétiques de la taille de l’homme à l’âge adulte, caractère multigénique s’il en est puisque presque 700 variants qui l’influencent ont déjà été définis, et montre que des variants rares dans des séquences codantes ont un impact plus important que la plupart de ceux répertoriés jusqu’ici (et qui sont en majorité des variants fréquents dans des séquences non codantes). Peut-on ainsi espérer résoudre l’irritant problème de l’héritabilité manquante (missing heritability), auquel deux chroniques ont été consacrées par le passé [2, 3] (→), et qui se manifeste dans ce cas par le fait que les 697 variants connus ne rendent compte à eux tous que de 20 % environ de l’héritabilité de la taille [4] ? Avant d’examiner les conclusions de cet article, il est nécessaire de faire un point d’ensemble sur les différentes classes de variants répertoriés dans le génome humain, à la lumière des études de séquençage à grande échelle qui ont été menées ces dernières années et qui ont largement précisé ce que certains appellent le « variome » humain1.
(→) Voir les Chroniques génomiques de B. Jordan, m/s n° 5, mai 2010, page 541, et m/s n° 3, mars 2011, page 323
Projet 1 000 génomes : état des lieux
Ce projet, dont le rapport final a été publié fin 2015 [5], a en fait établi les séquences d’ADN de 2 504 personnes appartenant à 26 populations correspondant aux différentes régions du globe, et a ainsi repéré près de 85 millions de SNP (single nucleotide polymorphisms) ainsi que 3,6 millions de courtes insertions ou délétions (indels) et 60 000 variants de structure (grandes insertions, délétions ou inversions). Je me concentre par la suite sur les SNP, seules variations qui puissent être rapidement étudiées à grande échelle grâce aux techniques de microarrays (ou « puces à ADN »). La base de données officielle dbSNP2 contient actuellement près de 150 millions de SNP, à l’époque (en 2015) 100 millions : il est clair que plus l’on séquence d’ADN humains, plus on va découvrir de variations, mais la plupart d’entre elles seront rares, présentes seulement chez un ou quelques individus. Sur les 85 millions de SNP trouvés dans l’étude 1 000 genomes, 64 millions sont rares, avec une fréquence de l’allèle mineur (MAF, pour minor allele frequency) inférieure à 0,5 % : l’allèle majeur a donc une fréquence d’au moins 99,5 %3 ; 12 millions environ sont peu fréquents (low frequency), avec une fréquence de l’allèle mineur comprise entre 0,5 et 5 %, 8 millions seulement sont fréquents (allèle mineur supérieur à 5 %). Cette prédominance des variants rares n’est apparente que lorsqu’on considère l’ensemble des génomes : si l’on examine un génome individuel, les proportions sont inversées. La séquence d’un génome typique présente 4 à 5 millions de différences avec la séquence de référence4, et la grande majorité de ces différences correspond à des variations fréquentes, seules 50 à 200 000 d’entre elles sont des variants rares (MAF inférieur à 0,5 %). En gros, et comme discuté précédemment [6] (→), les variants fréquents correspondent à des mutations anciennes et les variants rares à des substitutions plus récentes (100 à 300 générations d’après [5]).
(→) Voir la Chronique génomique de B. Jordan, m/s n° 4, avril 2014, page 463
Les limites des GWAS (genome-wide association studies) – et comment les contourner
Pour des raisons techniques et financières, l’essentiel des études d’association génome entier (GWAS) a été effectué en employant des microarrays pour repérer les allèles de SNP présents dans les ADN étudiés. Il faut en effet étudier des dizaines ou même des centaines de milliers de personnes pour repérer les associations génétiques de manière statistiquement significative, et même au tarif actuel de 1 000 à 2 000 dollars par génome, le séquençage d’un tel nombre d’échantillons aurait un coût prohibitif, sans parler des problèmes de stockage et de traitement de l’information. Les microarrays (puces à ADN) sont aujourd’hui bien plus abordables (de l’ordre de 50 dollars pièce), leur mise en œuvre est relativement simple et largement automatisée. Mais leur capacité est limitée, et, malgré une miniaturisation très poussée, ils ne permettent en général de définir qu’un million d’allèles de SNP dans l’ADN étudié – ce qui est déjà une belle performance ! Les microarrays employés pour les études GWAS vont donc généralement être ciblés sur les SNP les plus fréquents : ils auront ainsi le maximum de chances de détecter des différences entre individus au sein de leur ADN. On peut néanmoins se demander si ces différences sont les plus significatives du point de vue fonctionnel, et vouloir explorer au moins en partie l’univers des variants rares : c’est ce qu’ont tenté les auteurs de l’article cité au début [1]. Ils ont choisi d’utiliser un microarray commercial appelé ExomeChip et qui, comme son nom l’indique, détecte essentiellement des variants situés à l’intérieur de séquences codantes (dont l’ensemble constitue l’exome) et qui sont en majorité rares ou peu fréquents, avec une fréquence de l’allèle mineur inférieure à 5 %. On peut espérer que ces deux caractéristiques augmentent leurs chances de trouver des variants dont l’effet sur la taille soit relativement important. L’étude est pratiquée sur un très grand nombre de personnes, plus de 700 000, et implique de nombreux laboratoires et plus de cinq cents auteurs – c’est en fait une méta-analyse rassemblant différentes études d’association utilisant le même microarray. Elle est menée en deux étapes, « découverte » sur environ 450 000 participants, puis validation sur un échantillon indépendant de 250 000 personnes. Au total, il en résulte 83 variants confirmés dont 32 correspondent à des variations rares (MAF inférieur à 1 %, noter que cette définition de « rare » est moins restrictive que celle de 1 000 genomes [5]) et 51 à des faibles fréquences (MAF compris entre 1 % et 5 %).
On peut alors s’intéresser à la relation entre l’effet de chaque variant (la différence moyenne de taille observée selon que l’allèle majeur ou mineur est présent) et son degré de rareté (la fréquence de l’allèle mineur, MAF). Comme l’indique la Figure 1 , on constate que plus l’allèle mineur est rare, plus son effet sur la taille est important : la corrélation est frappante, et l’effet sur la taille de l’allèle rare de STC2 (Stanniocalcin 2) (MAF = 0,1 %, nous y reviendrons par la suite) atteint deux centimètres – c’est le maximum. Pour la plupart des variants plus fréquents (MAF supérieur à 5 %), l’effet tombe à quelques millimètres.
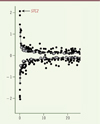 |
Figure 1. Effet de la présence de l’allèle mineur sur la taille (ordonnée, en cm) en fonction de la fréquence de cet allèle (MAF [minor allele frequency], abscisse, en pour cent). Selon les cas, l’allèle mineur a un effet positif ou négatif sur la taille ; les différents cercles correspondent à des variants détectés dans différentes phases de l’étude. Le gène STC2 (Stanniocalcin 2) est discuté dans le texte (extrait remanié de la figure 1 de [1]). |
Examinons, par exemple, le cas du gène STC2. Il n’avait jusqu’ici pas été impliqué dans la régulation de la croissance humaine, mais il a été montré, chez la souris, que son produit inhibe la protéinase PAPP-A (pregnancy-associated plasma protein-A). Cette dernière libère le facteur de croissance IGF-1 (insulin-like growth factor-1) en clivant la protéine IGFBP-4 (insulin growth factor binding protein-4). La surexpression de la protéine STC2 par un transgène introduit chez la souris augmente l’inhibition de PAPP-A et diminue la production d’IGF-1, avec comme résultat un phénotype de nanisme sévère [7]. Pour élucider ce qui se passe chez l’homme, les auteurs ont mené, sur des cultures de cellules humaines portant l’un ou l’autre allèle de STC2, une série d’études fonctionnelles. Celles-ci ont montré que l’allèle mineur de STC2 révélé par cette étude est exprimé au même niveau que l’allèle majeur, mais que la fixation de la protéine correspondante5 à PAPP-A et donc l’inhibition du clivage d’IGFBP-4 étaient diminués. Le résultat est ainsi une production plus importante du facteur IGF-1, qui est très vraisemblablement la cause de l’augmentation de la taille à l’âge adulte observée. On a donc bouclé la boucle, repéré par une analyse GWAS – ciblée sur les variants rares dans les séquences codantes – une variation associée à une augmentation relativement importante de la taille, et élucidé au niveau moléculaire le mécanisme probable de cet effet. Notons néanmoins que cette variation génique, une de celles qui entraîne l’effet le plus important sur la taille (Figure 1), n’ajoute en moyenne que deux centimètres à la stature de ses porteurs : cela relativise les fantasmes de fabrication d’individus de haute taille par genome editing sur l’embryon… [8] (→).
(→) Voir la Chronique de B. Jordan, m/s n° 6–7, juin-juillet 2015, page 691
Une moisson de résultats… et une interrogation persistante
Ce n’est bien sûr pas le seul résultat de cet article, qui identifie au total près de 100 variants nouveaux influençant la taille humaine, dont 24 pour lesquels l’effet atteint ou dépasse un centimètre. Certains d’entre eux se situent dans des gènes déjà impliqués dans des affections monogéniques touchant à la croissance du squelette et indiquent donc la présence, à côté de mutations pathogènes, de variants dans les mêmes gènes affectant de manière beaucoup moins drastique la croissance ; d’autres révèlent des cascades de signalisation dont le rôle dans la croissance humaine était jusque-là inconnu. De façon plus générale, l’implication de variants rares et peu fréquents dans la composante génétique de la taille humaine est bien mise en évidence par ce travail. Il n’en reste pas moins qu’à l’issue de cet examen de près de 250 000 SNP (le contenu de l’ExomeChip) chez plus de 700 000 personnes, l’ensemble de tous les variants repérés (passés et présents) n’explique au total que 27,4 % de l’héritabilité de la taille [1]. Grâce à ce travail considérable qui a impliqué des centaines de chercheurs, on est passé d’environ 20 % à près de 30 %, un progrès certes important mais qui n’en laisse pas moins plus de 70 % d’héritabilité non expliquée : le mystère de l’héritabilité manquante n’a pas disparu. Une solution à cette irritante question avait été proposée par l’équipe d’Eric Lander il y a quelques années [9] : l’évaluation de l’héritabilité serait faussée du fait que certains des locus génétiques impliqués sont en interaction (par exemple, parce qu’ils correspondent à différents éléments de la même cascade de régulation). Ces auteurs montrent que les interactions génétiques produisent une « héritabilité fantôme » (phantom heritability) qui peut rendre compte de la majeure partie de l’héritabilité manquante. Cette explication séduisante semble difficile à démontrer de manière expérimentale, et les mêmes auteurs, dans un article plus récent, indiquent qu’elle n’apporte probablement qu’une explication partielle [10]. C’est sans doute du côté des variants rares que se trouve la réponse. L’exploration à l’aide de l’ExomeChip ne peut par la force des choses qu’être très partielle puisqu’elle ne visualise qu’un petit sous-ensemble des variants rares repérés dans notre génome : si l’on veut aller plus loin, il faudra en passer par le séquençage. Celui-ci devra être pratiqué dans des conditions bien précises pour espérer tirer des conclusions solides : selon une analyse émanant elle aussi de l’équipe d’Eric Lander [10], il faudra travailler au niveau des exomes (pour des questions de coût, mais aussi de puissance statistique) et étudier quelques dizaines de milliers d’échantillons. Le jeu en vaut-il la chandelle ? Il ne sera peut-être pas nécessaire de lancer pour cela des programmes spécifiques. Le séquençage systématique d’ADN humains continue à se développer à grande vitesse, tout comme les efforts pour rattacher le maximum d’informations phénotypiques et cliniques à chacune des séquences obtenues : l’exploitation de ces grands ensembles de données devrait permettre à terme de répondre à de nombreuses questions de génétique et notamment à celle de l’architecture génétique des maladies ou caractères complexes.
Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Rappelons que les SNP sont pratiquement tous bialléliques, même si théoriquement quatre allèles sont possibles. Cela provient du fait qu’ils résultent d’une mutation en un point donné de l’ADN.
Qui porte une substitution d’un acide aminé.
Références
- Marouli E, Graff M, Medina-Gomez C, et al. Rare and low-frequency coding variants alter human adult height. Nature 2017 ; 542 : 186–190. [CrossRef] [PubMed] [Google Scholar]
- Jordan B. À la recherche de l’héritabilité perdue Med Sci (Paris) 2010 ; 26 : 541–543. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Jordan B. Maladie de Crohn et GWAS, d’analyses en méta-analyses. Med Sci (Paris) 2011 ; 27 : 323–325. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Wood AR, Esko T, Yang J, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 2014 ; 46 : 1173–1186. [Google Scholar]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015; 526 : 68–74. [CrossRef] [PubMed] [Google Scholar]
- Jordan B. Variants rares et explosion démographique. Med Sci (Paris) 2014 ; 30 : 463–466. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Jepsen MR, Kløverpris S, Mikkelsen JH, et al. Stanniocalcin-2 inhibits mammalian growth by proteolytic inhibition of the insulin-like growth factor axis. J Biol Chem 2015 ; 290 : 3430–3439. [CrossRef] [PubMed] [Google Scholar]
- Jordan B. Thérapie génique germinale, le retour ?. Med Sci (Paris) 2015 ; 31 : 691–695. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci USA 2012 ; 109 : 1193–1198. [CrossRef] [Google Scholar]
- Zuk O, Schaffner SF, Samocha K, et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci USA 2014 ; 111 : E455–E464. [CrossRef] [Google Scholar]
Liste des figures
|
Figure 1. Effet de la présence de l’allèle mineur sur la taille (ordonnée, en cm) en fonction de la fréquence de cet allèle (MAF [minor allele frequency], abscisse, en pour cent). Selon les cas, l’allèle mineur a un effet positif ou négatif sur la taille ; les différents cercles correspondent à des variants détectés dans différentes phases de l’étude. Le gène STC2 (Stanniocalcin 2) est discuté dans le texte (extrait remanié de la figure 1 de [1]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.