")
")
| Issue |
Med Sci (Paris)
Volume 32, Number 12, Décembre 2016
|
|
|---|---|---|
| Page(s) | 1068 - 1071 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20163212008 | |
| Published online | 03 January 2017 | |
Rôle des récepteurs NMDA endothéliaux dans un modèle murin de sclérose en plaques
Role of endothelial NMDA receptors in a mouse model of multiple sclerosis
1
Normandie Univ, Unicaen, Inserm, physiopathology and imaging of neurological disorders (PhIND), boulevard Becquere, 14000 Caen, France
2
Service de biologie, délégation recherche clinique et innovation, centre hospitalier universitaire de Caen, 14000 Caen, France
La sclérose en plaques (SEP) est une maladie auto-immune du système nerveux central conduisant à la destruction de la myéline isolant les fibres nerveuses. Cette maladie est la première cause de handicap chez le jeune adulte et touche actuellement environ 90 000 personnes en France. Grâce aux modèles animaux de SEP, la compréhension de la physiopathologie de cette maladie, bien que toujours incomplète, a largement progressé au cours des dernières décennies. Aujourd’hui, on considère qu’une altération locale des barrières hématoencéphalique1 (dans le cerveau) et hématomédullaire1 (dans la moelle épinière) est associée à l’apparition de la maladie, conduisant à l’infiltration de cellules immunitaires vers les tissus nerveux, puis à la destruction de la myéline et des axones.
Diverses études ont mis en évidence les effets toxiques du glutamate dans les modèles animaux de SEP [1]. Ces résultats ont conduit à la mise en place d’essais cliniques, dont certains sont toujours en cours, visant à tester l’efficacité de modulateurs des récepteurs du glutamate, et plus particulièrement du récepteur du N-méthyl-D-aspartate (NMDA), dans la SEP [1]. Au-delà de son expression classiquement décrite au niveau des neurones, où il participe à la neurotransmission glutamatergique [2] (→), le récepteur NMDA est aussi exprimé par de nombreux autres types cellulaires, en particulier les cellules endothéliales cérébrales [3]. Il a de plus été suggéré que le récepteur NMDA pouvait intervenir dans le contrôle de la migration des cellules inflammatoires à travers l’endothélium vasculaire cérébral [3].
(→) Voir la Synthèse de M. Gielen, m/s n° 1, janvier 2010, page 65
Le récepteur NMDA présente plusieurs sites régulateurs, dont un site sensible à une protéase, l’activateur tissulaire du plasminogène (tPA). L’interaction du tPA avec les récepteurs NMDA induit une augmentation d’activité de ces récepteurs [4, 5]. Notre équipe a montré que le tPA, en activant les récepteurs NMDA des cellules endothéliales cérébrales, permettait l’infiltration des cellules inflammatoires vers le tissu cérébral et la moelle épinière [6]. Afin de bloquer l’action du tPA sur ce récepteur, nous avons développé un anticorps monoclonal (Glunomab®) [7] reconnaissant le site de fixation du tPA sur la partie N-terminale du récepteur NMDA (Figure 1). En traitant des souris avec le Glunomab®, nous avons arrêté la progression des dommages chez les souris soumises à un modèle de SEP [6].
 |
Figure 1. Le Glunomab® cible un site régulateur du récepteur NMDA activé par le tPA. A. Représentation schématique du récepteur NMDA. Le récepteur NMDA contient la sous-unité GluN1, comportant un site de fixation de la glycine (co-agoniste obligatoire, rond vert) et un site de fixation et d’action du tPA (étoile bleue). La sous-unité GluN2 contient, elle, le site de fixation du glutamate (agoniste principal, rond orange). B. Fixation du Glunomab®. Le Glunomab® se fixe sur le site de fixation du tPA, situé au niveau du domaine N-terminal de la sous-unité GluN1. C. Détail du site de fixation du Glunomab® (en gras : épitope minimal du tPA ; en orange : la lysine 178, site de clivage de GluN1 par le tPA). NMDA : N-méthyl-D-aspartate ; tPA : activateur tissulaire du plasminogène. |
Le Glunomab® agit sur la barrière hématomédullaire pour bloquer l’infiltration des cellules inflammatoires
L’infiltration des cellules inflammatoires à travers les barrières hématoencéphalique et hématomédullaire peut être mimée en utilisant un modèle de culture de cellules endothéliales. Dans ce modèle, les monocytes et les lymphocytes sont capables de traverser une couche de cellules endothéliales cérébrales humaines (cellules hMEC-D3 [8]) en conditions inflammatoires. En traitant les cellules endothéliales avec le Glunomab®, nous avons bloqué cette infiltration. Avant de passer à des tests chez l’animal, nous avons préalablement étudié si le Glunomab® avait la capacité de cibler les cellules endothéliales de la moelle épinière. Nous avons observé sur des coupes de moelle épinière de souris et de sujets humains que le Glunomab® se fixait au niveau des vaisseaux de la moelle épinière, à leur surface luminale et à proximité des protéines formant les jonctions serrées (claudine 5, occludine et zonula occludens-1 [ZO-1]), nécessaires à la perméabilité de la barrière hématomédullaire. Ces résultats montraient, d’une part, pour la première fois, la présence de récepteurs NMDA au niveau de l’endothélium vasculaire de la moelle épinière et suggéraient, d’autre part, que le Glunomab® puisse agir sur ces récepteurs NMDA.
Le Glunomab® bloque la progression de la maladie dans un modèle de SEP chez la souris
Le Glunomab® étant capable de bloquer l’infiltration des cellules immunitaires dans des modèles cellulaires et de se fixer aux récepteurs NMDA exprimés à la surface des vaisseaux de la moelle épinière, nous avons par la suite testé son efficacité dans un modèle de SEP chez la souris : l’encéphalite auto-immune expérimentale (EAE).
Dans un premier temps, nous avons réalisé trois injections successives de notre anticorps, par voie intraveineuse chez la souris : (1) à la déclaration des symptômes, (2) au cours de la phase de progression de ces symptômes et (3) au cours de la phase chronique. Ces injections ont permis d’infléchir la courbe de progression des symptômes : les animaux traités avec un anticorps contrôle ont subi une aggravation des symptômes jusqu’à la paralysie des membres postérieurs, alors que, chez ceux traités avec le Glunomab®, ces symptômes se sont limités à une faiblesse des membres postérieurs. Par la suite, une deuxième phase de tests a été réalisée avec une seule injection chez des animaux présentant une faiblesse avérée des membres postérieurs. De nouveau, alors que les animaux contrôles évoluaient jusqu’à la paralysie, la progression des symptômes a été totalement bloquée par l’injection de Glunomab®. En procédant à l’analyse des tissus de moelle épinière, nous avons mis en évidence une réduction très importante de l’étendue des lésions démyélinisantes chez les souris traitées avec le Glunomab®, ce qui concorde avec leurs symptômes moins graves.
Nous avons ensuite cherché à comprendre si cet effet bénéfique du Glunomab® était lié à une action sur la barrière hématomédullaire. Nous avons observé par des techniques d’immunohistologie que VCAM-1 (vascular cell adhesion protein 1), un marqueur d’activation endothéliale, était moins présent à la surface des vaisseaux chez les souris traitées avec le Glunomab® que chez les souris contrôles. De plus, le fibrinogène, qu’on ne retrouve dans la moelle épinière que si la barrière hématomédullaire est altérée, était également moins présent chez les souris traitées avec l’anticorps que chez les souris contrôles. Enfin, nous avons mis en évidence par cytométrie en flux et immunohistochimie une réduction drastique de l’infiltration des cellules immunitaires (lymphocytes T CD4+, monocytes/macrophages, neutrophiles et cellules dendritiques) dans la moelle épinière des souris traitées avec le Glunomab®.
Conclusions et perspectives
Nos travaux montrent que le tPA, en augmentant l’activité des récepteurs NMDA présents sur les cellules endothéliales, facilite l’infiltration des cellules immunitaires et inflammatoires. Le Glunomab®, en empêchant l’action du tPA sur ces récepteurs NMDA, bloque l’infiltration des cellules immunitaires et inflammatoires, et est ainsi capable de limiter la progression des symptômes dans un modèle de SEP chez la souris (Figure 2). Ces résultats suggèrent que le site d’activation par le tPA sur les récepteurs NMDA des vaisseaux pourrait constituer une cible thérapeutique d’intérêt dans la SEP. Un anticorps-médicament, sur le modèle du Glunomab®, pourrait donc présenter des effets bénéfiques contribuant à limiter la progression des symptômes dans cette maladie.
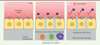 |
Figure 2. Mode d’action du Glunomab®. A. Les cellules endothéliales cérébrales présentent à leur surface apicale (face en contact avec le sang) des récepteurs NMDA. B. En cas d’activation de ces récepteurs par le tPA, la barrière hématomédullaire s’ouvre, permettant le passage de cellules inflammatoires (lymphocytes, neutrophiles et monocytes) vers le tissu nerveux, causant ainsi les dommages à ce tissu. C. Le Glunomab®, en empêchant la fixation du tPA sur le récepteur NMDA, limite l’entrée des cellules inflammatoires et réduit ainsi les dommages. NMDA : N-méthyl-D-aspartate ; tPA : activateur tissulaire du plasminogène. |
En marge des applications thérapeutiques potentielles, notre étude montre également l’implication de ces récepteurs NMDA endothéliaux dans la régulation de la perméabilisation des barrières hématoencéphalique et hématomédullaire. Toutefois, le mécanisme précis conduisant à une « ouverture » de ces barrières à la suite de l’activation des récepteurs NMDA reste à définir, même si plusieurs hypothèses ont été formulées dans des études antérieures, dont l’inhibition d’expression de protéines des jonctions serrées [9] ou l’induction d’une toxicité mitochondriale [10]. Il conviendra également, en marge de leurs rôles au cours de pathologies telles que la SEP, de mieux comprendre comment les récepteurs NMDA endothéliaux agissent en conditions physiologiques.
Une autre question intéressante est celle des ligands endogènes qui permettent d’activer les récepteurs NMDA endothéliaux. Différents types de cellules immunitaires et inflammatoires produisent du glutamate, agoniste naturel du récepteurs NMDA [1], mais également du tPA [11, 12], qui augmente l’activité de ce récepteur. On peut donc imaginer que ces cellules, en produisant du glutamate et du tPA, vont participer à l’ouverture des barrières hématoencéphalique et hématomédullaire, pour faciliter leur propre infiltration vers les tissus nerveux. Le fait que le Glunomab® bloque leur infiltration, comme nous le décrivons dans notre étude, est en faveur de cette hypothèse.
Certains types d’auto-anticorps dirigés contre les récepteurs NMDA sont responsables d’une maladie neurologique nommée encéphalite anti-récepteurs NMDA [13]. Toutefois, ces auto-anticorps ciblent une région différente (acides aminés 368 et 369) [14] de celle reconnue par le Glunomab® (acides aminés 169 à 192). De plus, à la différence des anticorps responsables de l’encéphalite [15], le Glunomab® ne réduit pas l’activité basale des récepteurs NMDA, mais seulement l’augmentation de sa réponse par le tPA, ce qui va dans le sens d’une absence d’effet sur la transmission glutamatergique physiologique. Les effets secondaires inacceptables des composés bloquant totalement les récepteurs NMDA, ont contraint à arrêter les essais cliniques [1]. Il y a donc un intérêt grandissant pour des molécules à action modulatrice des récepteurs NMDA, qui n’affecteraient pas la transmission basale. Le Glunomab®, qui module spécifiquement le site d’action du tPA sur ce récepteur, entre dans cette catégorie de modulateurs et apporte donc de nouveaux espoirs de translation vers la clinique.
Liens d’intérêt
Les auteurs de cet article apparaissent comme inventeurs sur un brevet (ref : WO/2014/187879) concernant l’utilisation du Glunomab® dans les maladies neurodégénératives.
Les barrières hématoencéphalique (dans le cerveau) et hématomédullaire (dans la moelle épinière) sont des barrières physiologiques limitant le passage de molécules et de cellules depuis le sang vers les tissus nerveux. Les cellules endothéliales formant ces barrières ont la particularité d’être jointes par des jonctions serrées et de ne pas présenter de fenestrations. De plus, des prolongements (« pieds ») astrocytaires et des péricytes viennent entourer ces vaisseaux pour augmenter encore leur imperméabilité.
Références
- Macrez R, Stys PK, Vivien D, et al. Mechanisms of glutamate toxicity in multiple sclerosis: biomarker and therapeutic opportunities. Lancet Neurol 2016 ; 15 : 1089–1102. [CrossRef] [PubMed] [Google Scholar]
- Gielen M. Fonctionnement des récepteurs-canaux du glutamate : des proteines responsables de la transmission synaptique excitatrice. Med Sci (Paris) 2010 ; 26 : 65–72. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Reijerkerk A, Kooij G, van der Pol SMA, et al. The NR1 subunit of NMDA receptor regulates monocyte transmigration through the brain endothelial cell barrier. J Neurochem 2010 ; 113 : 447–453. [CrossRef] [PubMed] [Google Scholar]
- Nicole O, Docagne F, Ali C, et al. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med 2001 ; 7 : 59–64. [CrossRef] [PubMed] [Google Scholar]
- Samson AL, Medcalf RL Tissue-type plasminogen activator: a multifaceted modulator of neurotransmission and synaptic plasticity. Neuron 2006 ; 50 : 673–678. [CrossRef] [PubMed] [Google Scholar]
- Macrez R, Ortega MC, Bardou I, et al. Neuroendothelial NMDA receptors as therapeutic targets in experimental autoimmune encephalomyelitis. Brain 2016 ; 139 : 2406–2419. [CrossRef] [PubMed] [Google Scholar]
- Lesept F, Chevilley A, Jezequel J, et al. Tissue-type plasminogen activator controls neuronal death by raising surface dynamics of extrasynaptic NMDA receptors. Cell death Dis 2016 ; doi: 10.1038/cddis.2016.279 [Google Scholar]
- Weksler BB, Subileau EA, Perrière N, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J 2005 ; 19 : 1872–1874. [Google Scholar]
- András IE, Deli MA, Veszelka S, et al. The NMDA and AMPA/KA receptors are involved in glutamate-induced alterations of occludin expression and phosphorylation in brain endothelial cells. J Cereb Blood Flow Metab 2007 ; 27 : 1431–1443. [CrossRef] [PubMed] [Google Scholar]
- Kamat PK, Kalani A, Tyagi SC, et al. Hydrogen sulfide epigenetically attenuates homocysteine-induced mitochondrial toxicity mediated through NMDA receptor in mouse brain endothelial (bEnd3) cells. J Cell Physiol 2015 ; 230 : 378–394. [CrossRef] [PubMed] [Google Scholar]
- Lin L, Jin Y, Mars WM, et al. Myeloid-derived tissue-type plasminogen activator promotes macrophage motility through FAK, Rac1, and NF-κB pathways. Am J Pathol 2014 ; 184 : 2757–2767. [CrossRef] [PubMed] [Google Scholar]
- Uhl B, Zuchtriegel G, Puhr-Westerheide D, et al. Tissue plasminogen activator promotes postischemic neutrophil recruitment via its proteolytic and nonproteolytic properties. Arterioscler Thromb Vasc Biol 2014 ; 34 : 1495–1504. [CrossRef] [PubMed] [Google Scholar]
- Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008 ; 7 : 1091–1098. [CrossRef] [PubMed] [Google Scholar]
- Gleichman AJ, Spruce LA, Dalmau J, et al. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci 2012 ; 32 : 11082–11094. [CrossRef] [PubMed] [Google Scholar]
- Mikasova L, De Rossi P, Bouchet D, et al. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain 2012 ; 135 : 1606–1621. [CrossRef] [PubMed] [Google Scholar]
© 2016 médecine/sciences – Inserm
Liste des figures
|
Figure 1. Le Glunomab® cible un site régulateur du récepteur NMDA activé par le tPA. A. Représentation schématique du récepteur NMDA. Le récepteur NMDA contient la sous-unité GluN1, comportant un site de fixation de la glycine (co-agoniste obligatoire, rond vert) et un site de fixation et d’action du tPA (étoile bleue). La sous-unité GluN2 contient, elle, le site de fixation du glutamate (agoniste principal, rond orange). B. Fixation du Glunomab®. Le Glunomab® se fixe sur le site de fixation du tPA, situé au niveau du domaine N-terminal de la sous-unité GluN1. C. Détail du site de fixation du Glunomab® (en gras : épitope minimal du tPA ; en orange : la lysine 178, site de clivage de GluN1 par le tPA). NMDA : N-méthyl-D-aspartate ; tPA : activateur tissulaire du plasminogène. |
| Dans le texte | |
|
Figure 2. Mode d’action du Glunomab®. A. Les cellules endothéliales cérébrales présentent à leur surface apicale (face en contact avec le sang) des récepteurs NMDA. B. En cas d’activation de ces récepteurs par le tPA, la barrière hématomédullaire s’ouvre, permettant le passage de cellules inflammatoires (lymphocytes, neutrophiles et monocytes) vers le tissu nerveux, causant ainsi les dommages à ce tissu. C. Le Glunomab®, en empêchant la fixation du tPA sur le récepteur NMDA, limite l’entrée des cellules inflammatoires et réduit ainsi les dommages. NMDA : N-méthyl-D-aspartate ; tPA : activateur tissulaire du plasminogène. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.