")
")
| Issue |
Med Sci (Paris)
Volume 30, Number 8-9, Août–Septembre 2014
|
|
|---|---|---|
| Page(s) | 758 - 764 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20143008013 | |
| Published online | 01 September 2014 | |
Les infections bactériennes vues du génome eucaryote
Cassures double-brin, inflammation et cancer
Bacterial infections as seen from the eukaryotic genome: DNA double strand breaks, inflammation and cancer
1
Inserm UMR_S 1038, biologie à grande échelle (BGE), 38054
Grenoble, France
2
CEA, institut de recherches en technologies et sciences pour le vivant (iRTSV), BEG, 38054
Grenoble, France
3
Université Grenoble-Alpes, 38000
Grenoble, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Un nombre croissant d’études indiquent que l’infection par des bactéries pathogènes induit de graves lésions sur le génome de l’hôte, notamment des cassures double-brin. Même après réparation de l’ADN, il reste souvent, au niveau chromosomique, des séquelles qui peuvent générer une instabilité génétique lors d’une division ultérieure. L’inflammation chronique favorise l’expansion des bactéries génotoxiques dans le microbiote intestinal et l’apparition de carcinomes du côlon. Les bactéries manipulent les points de contrôle du cycle cellulaire et les voies de réparation de l’ADN, mais les acteurs moléculaires induisant les lésions sur l’ADN ne sont pas identifiés avec certitude, en dépit de fortes présomptions sur les espèces réactives de l’oxygène.
Abstract
An increasing number of studies report that infection by pathogenic bacteria alters the host genome, producing highly hazardous DNA double strand breaks for the eukaryotic cell. Even when DNA repair occurs, it often leaves “scars” on chromosomes that might generate genomic instability at the next cell division. Chronic intestinal inflammation promotes the expansion of genotoxic bacteria in the intestinal microbiote which in turn triggers tumor formation and colon carcinomas. Bacteria act at the level of the host DNA repair machinery. They also highjack the host cell cycle to allow themselves time for replication in an appropriate reservoir. However, except in the case of bacteria carrying the CDT nuclease, the molecular mechanisms responsible for DNA lesions are not well understood, even if reactive oxygen species released during infection make good candidates.
© 2014 médecine/sciences – Inserm
Le maintien de l’intégrité du génome est un processus vital pour assurer la viabilité et le renouvellement cellulaires. Les lésions de l’ADN peuvent aller de la simple modification chimique d’une base de l’ADN à des cassures sur un brin ou simultanément sur les deux brins de la double hélice [1]. Les origines des dommages sont variées ; par exemple physiologiques et programmés, certains sont inhérents à un défaut lors de la réplication de l’ADN, d’autres encore surviennent en présence d’agents génotoxiques externes (radiations, chimiothérapies, UV, espèces réactives de l’oxygène [ROS], virus, etc.). Depuis quelques années, il devient évident que certaines infections bactériennes altèrent l’ADN des cellules infectées. Historiquement, le cas d’Helicobacter pylori a permis de défricher le terrain, puis la découverte de la toxine colibactine, en 2006 chez Escherichia coli, a contribué à des avancées majeures dans ce domaine. En à peine quelques années, la génotoxicité d’une dizaine d’autres bactéries a été révélée. Ce champ d’investigation est en pleine expansion, et les résultats récents confirment désormais le lien entre infection bactérienne et cancer ( Tableau I ).
Liens entre infection bactérienne et cancer.
Les cassures double-brin de l’ADN, leur signalisation et leur réparation
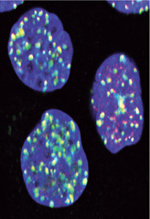
Bien qu’elles ne représentent qu’un petit nombre des dommages observés lors d’un stress génotoxique, les cassures double-brin constituent les lésions biologiques les plus toxiques pour la cellule [2]. Une seule lésion non réparée peut conduire à la mort cellulaire ; une réparation imparfaite peut induire une instabilité chromosomique et des remaniements chromosomiques. La détection et la signalisation des lésions double-brin se font par l’intermédiaire d’un réseau de protéines impliquant une cascade de phosphorylations, puis une cascade d’ubiquitinations, le tout rassemblé dans un macro-complexe appelé IRIF (ionizing radiation induced foci) (Figure 1). Un élément central est la phosphorylation de l’histone H2AX sur la sérine 139 (γH2AX) par la kinase ATM (ataxia telangiectasia mutated), ce qui conduit à l’activation de p53 et des points d’arrêt du cycle cellulaire [2]. En fonction des dommages engendrés, la cellule peut entrer en apoptose, réparer son ADN ou entrer dans une phase de sénescence (Figure 1B). Deux mécanismes permettent la réparation des cassures double-brin. En bordure du point de contrôle S/G2 du cycle cellulaire, alors que l’ADN a été récemment répliqué, la voie de recombinaison homologue (HR) est activée de préférence. La réparation par HR est fidèle, sans perte d’informations, en raison de la présence d’un brin matrice original à copier. Le mécanisme de NHEJ (non homologous end joining) est mis en œuvre dans les autres configurations du cycle cellulaire. N’ayant pas de brin matrice à copier, la réparation par NHEJ se fait avec une perte d’information ; elle est potentiellement mutagène.
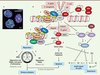 |
Figure 1. Conséquences des cassures double-brin de l’ADN et mécanismes de réparation. A. Les foyers IRIF marquant les cassures double-brin. Lors d’un stress génotoxique provoquant des cassures double-brin de l’ADN, l’histone H2AX est phosphorylée sur la sérine en position 139 (γH2AX) en quelques minutes. De nombreuses molécules γH2AX s’accumulent sur des centaines de kilobases de part et d’autres des cassures double-brin, formant des foyers nucléaires appelés IRIF (ionizing radiation-induced foci). Chaque foyer témoigne d’une cassure et est constitué d’un macro-complexe de protéines de signalisation et de réparation. En rouge γH2AX, en vert 53BP1, en bleu l’ADN. Le jaune représente la superposition des marquages rouge et vert. Échelle : 10 µm. B. Détection et signalisation des cassures double-brin de l’ADN. Une relaxation de la chromatine est initiée très rapidement au site de cassure de l’ADN (non représentée). Le senseur le plus proximal identifié est le complexe MNR, composé de Mre11, Nbs1 et Rad50, qui possède la capacité intrinsèque de lier l’ADN (étape 1). La kinase ataxia telangiectasia mutated (ATM), inactive sous forme de dimère (étape 2), est activée par autophosphorylation sur sa sérine 1981, et phosphoryle le complexe MNR, H2AX, MDC1 et p53 (étape 3). γH2AX constitue un site de liaison à haute affinité pour la protéine MDC1 qui, à son tour, orchestre le recrutement des autres effecteurs plus tardifs sur la chromatine endommagée. RNF8 et RNF168 sont deux ubiquitine-ligases qui ubiquitinent la chromatine et stimulent la fixation locale des protéines de réparation 53BP1 et BRCA1 (breast cancer 1) (étape 4). Les deux voies de réparation des cassures double-brin sont la recombinaison homologue (HR) et la voie NHEJ (non homologous end joining). La voie HR (étape 5) nécessite une matrice d’ADN dupliqué ; elle est donc active essentiellement à la phase S et G2 du cycle cellulaire. Elle assure une réparation sans erreur et sans perte d’information génétique. La voie NHEJ (étape 6) est active durant tout le cycle cellulaire et peut causer des pertes d’informations génétiques. Le cycle cellulaire est arrêté aux transitions G1/S ou G2/M, après activation de Chk1, Chk2 et Cdc25 (étape 7), donnant le temps aux mécanismes de réparation d’agir afin d’éviter la duplication du génome ou la division cellulaire alors que l’ADN est endommagé. L’apoptose dépendante de p53 intervient quand la cellule ne peut réparer son ADN (étape 8). Les points d’action des différentes bactéries citées dans le texte sont représentés. U : ubiquitine ; P : phosphorylation. |
Helicobacter pylori, un casseur connu de longue date !
Helicobacter pylori est une bactérie à Gram négatif colonisant l’estomac chez une personne sur deux. Si elle reste asymptomatique chez 80 % des individus infectés, les 20 % restants développent une gastrite chronique qui persiste toute la vie si l’infection n’est pas traitée. H. pylori est associée aux cancers gastriques, dans un contexte d’infection chronique et d’inflammation de la muqueuse gastrique. De nombreuses études ont montré que l’infection par H. pylori affectait le génome eucaryote et la conservation de son intégrité [3]. La bactérie induit une diminution de la synthèse des protéines réparant les mésappariements de l’ADN [4] et les excisions de base [5, 6], ce qui augmente le risque de mutation dans les cellules gastriques. De fait, la fréquence des mutations est plus élevée chez les souris infectées par H. pylori, avec une prévalence des mutations de AT vers CG, suggérant des dommages oxydatifs de l’ADN [7]. Umeda et al. ont découvert que CagA, un facteur de virulence délivré dans la cellule infectée [37], provoquait des retards lors de la prophase et de la métaphase, une mauvaise orientation du fuseau mitotique et un axe de division anormal, générant des anomalies de ségrégation des chromosomes et une instabilité chromosomique [8]. H. pylori serait également capable d’altérer le niveau du régulateur pro-apoptotique p53, en favorisant son ubiquitination puis sa dégradation par le protéasome, d’une façon dépendante de CagA [9, 10]. Même si H. pylori n’est pas un pathogène intracellulaire, son interaction avec la cellule hôte fournit un certain bénéfice pour la bactérie, ce qui contribuerait à l’établissement d’une infection chronique dans cette niche privilégiée en retardant la mort cellulaire [10].
Depuis 2011, on sait que l’infection par H. pylori provoque des cassures double-brin dans les cellules épithéliales gastriques. Des foyers nucléaires de type IRIF (γH2AX, 53BP1 [p53 binding protein 1] et MDC1 [mediator of DNA damage checkpoint 1]) se forment, d’une façon dépendante de la kinase ATM [11]. De grosses cassures au niveau des chromatides sont visibles sur des métaphases de cellules infectées, mais elles seraient indépendantes des facteurs de virulence connus de la bactérie. L’infection par H. pylori génère la production d’espèces réactives de l’oxygène et de l’azote (ROS) mais, malgré cette corrélation temporelle, la production de ROS ne serait pas nécessaire pour produire des cassures double-brin. L’éradication de H. pylori conduit à la réparation efficace des cassures double-brin et à la survie des cellules [11]. Cependant, si l’infection est prolongée, des cassures résiduelles, non réparées, persistent et affectent la viabilité cellulaire, probablement en raison de la saturation de la machinerie de réparation. L’induction de cassures double-brin, suivie d’une réparation imprécise, serait compatible avec les propriétés carcinogènes de H. pylori et contribuerait aux fréquentes anomalies chromosomiques observées dans les cancers gastriques.
Escherichia coli se fait génotoxique
Escherichia coli est la bactérie la plus abondante de la flore commensale de l’intestin où elle contribue à différentes fonctions telles que la dégradation des aliments [12], la modulation du système immunitaire ou encore la protection vis-à-vis de microorganismes pathogènes. La colonisation de l’intestin commence quelques jours après la naissance et persiste toute la vie. L’association avec l’hôte est une symbiose ; cependant il existe des souches pathogènes d’E. coli qui génèrent de multiples effets délétères au niveau intestinal. La pathogénicité des E. coli du groupe phylogénique B2 est souvent associée à la présence d’un îlot génomique appelé pks qui contient une vingtaine de gènes impliqués dans la biosynthèse de la colibactine, une génotoxine découverte en 2006 [13]. La structure de la colibactine, un composé hybride polykétide-peptide résultant des activités enzymatiques codées par l’îlot pks, n’est pas connue à ce jour.
Lorsque des souches porteuses de l’îlot pks infectent les cellules eucaryotes, des cassures double-brin de l’ADN ont lieu et la cascade de signalisation partant de la kinase ATM est activée [13] : phosphorylation d’ATM, d’H2AX et des effecteurs en aval tels que Chk2 (checkpoint kinase 2) et Chk1 (Figure 1B, étape 7). Chk1 et Chk2 phosphorylent alors la phosphatase Cdc25, la rendant inactive. Il en résulte un arrêt du cycle cellulaire à la transition G2/M ou G1/S. Ces cassures double-brin sont retrouvées in vivo dans les entérocytes de la muqueuse du côlon des souris infectées avec des E. coli pks+ [14]. À faible MOI (multiplicity of infection), les cellules réparent leur ADN et reprennent leur division, tandis que les cellules déficientes dans la voie de réparation NHEJ meurent massivement [14]. Cependant, certaines cellules gardent des foyers γH2AX comme des cicatrices de l’infection et des aberrations mitotiques et chromosomiques. L’infection peut aussi induire une aneuploïdie (gain ou perte de chromosomes) ou une tétraploïdie (cellule 8n), conséquences des anomalies de division. Enfin, il est observé une augmentation de la fréquence des mutations et une capacité accrue des cellules à proliférer en milieu semi-solide, signe d’un début de transformation [14]. Dans ces conditions, on comprend bien qu’au cours du temps, les multiples lésions induites par la colibactine s’accumulent, et qu’elles pourraient contribuer au processus de transformation cellulaire, une étape critique avant la cancérisation.
Lorsque l’infection est massive, les cellules subissent, en plus des cassures de leur ADN, un arrêt irréversible de leur cycle cellulaire, et elles s’engagent vers un état de sénescence dans lequel elles ne se divisent plus [15, 35], avec persistance possible de gros foyers γH2AX. Des ROS intracellulaires et mitochondriaux sont produits au bout de quelques jours et, de façon très intéressante, la sénescence peut se propager, in vitro, à des cellules non infectées par un effet de voisinage. Les ROS et les facteurs sécrétés seraient impliqués dans la transmission de l’information aux cellules adjacentes. Ceci diffère de H. pylori pour lequel un contact direct entre la cellule eucaryote et la bactérie est nécessaire [11]. La sénescence pourrait servir d’alternative fonctionnelle à l’apoptose, comme un dernier recours pour empêcher une cellule porteuse de lésions de se répliquer, se transformer et devenir cancéreuse, avant de former une tumeur.
Et quand tout un microbiote se déchaîne paisiblement, en toute harmonie
À l’échelle de l’organisme entier, des études récentes ont permis de mieux comprendre les infections bactériennes impliquant E. coli. Arthur et al. [16] ont utilisé un modèle inflammatoire de souris IL (interleukine) 10−/- dans lequel les animaux développent une intolérance à leur propre microbiote intestinal, une inflammation chronique et des colites. La comparaison du microbiote des souris sauvages avec celui des souris IL10−/- a révélé que les souris avec une inflammation hébergeaient une flore commensale enrichie en plusieurs groupes bactériens, à nombre constant de bactéries. Ainsi, la quantité des entérobactéries E. coli est multipliée par 100 ! Une corrélation entre la présence de cancers induits par l’azoxyméthane, un carcinogène provoquant des carcinomes du côlon, et l’état inflammatoire des souris a été mise en évidence [16]. Chez la souris, les bactéries qui expriment la colibactine (souche pks+) provoquent davantage de tumeurs du côlon et une invasion plus importante des tissus, qu’une souche isogénique sans colibactine, alors que les manifestations inflammatoires sont identiques dans les deux cas. Les souches d’E. coli pks+ sont sur-représentées sur des biopsies humaines de patients atteints de carcinomes du côlon (66,7 %) et de maladies inflammatoires intestinales (40 %) par rapport aux biopsies de patients n’ayant ni l’un ni l’autre de ces syndromes (20,8 %) [16]. Il semblerait donc que l’inflammation chronique de l’intestin, aussi bien chez la souris que chez l’homme, favorise l’expansion de bactéries avec un potentiel génotoxique. De plus, cet état inflammatoire créerait un environnement favorisant la cancérogenèse, en agissant aussi bien sur l’hôte que sur la composition du microbiote intestinal.
La mise en évidence des activités génotoxiques des bactéries pathogènes a eu le mérite de susciter une réflexion sur certaines stratégies thérapeutiques. Par exemple, la souche E. coli Nissle 1917, une souche commensale très bonne colonisatrice chez l’homme et la souris, a été très utilisée comme traitement probiotique dans les désordres intestinaux, tels que la maladie de Crohn ou la colite ulcéreuse, avant l’identification d’un îlot pks potentiellement dangereux dans son génome. Une étude a depuis montré que la présence de colibactine était indissociable de l’activité anti-inflammatoire de cette souche [17], et qu’il convenait d’évaluer le rapport bénéfice/risque de son utilisation à long terme.
L’îlot pks présent dans le génome d’entérobactéries autres que E. coli est lui aussi fonctionnel et générateur de cassures double-brin de l’ADN pour des souches de Citrobacter koseri, Enterobacter aerogenes et Klebsiella pneumoniae [18]. Chez la souris, l’infection par K. pneumoniae pks+ induit en un à deux jours des cassures double-brin de l’ADN dans les cellules parenchymateuses du foie, ceci à condition que la colibactine soit présente [19]. Or, dans la population de Taïwan, K. pneumoniae est le pathogène prédominant associé à l’abcès à pyogènes du foie, une infection sévère suspectée d’accroître le risque de développer secondairement un cancer colorectal [19]. Des études complémentaires seront nécessaires pour valider cette hypothèse.
S’il est établi que la colibactine induit de graves dommages à l’ADN, son mécanisme d’action n’est pas élucidé à ce jour. Ses effets pourraient être indirects ; par exemple elle induirait un stress oxydatif et la production de radicaux libres attaquant directement l’ADN, mais les ROS ne sont pas détectés immédiatement après l’infection [15]. Il faut noter que H. pylori induit la production de ROS, mais que le blocage de leur action n’empêche pas les cassures double-brin [11], ce qui suggère d’autres mécanismes qui nécessiteraient, peut-être, la présence de nucléase(s).
CDT, une ADNase bactérienne qui attaque le génome eucaryote
La toxine CDT (cytolethal distending toxin) est la première toxine identifiée comme bloquant le cycle cellulaire au stade G2/M [20]. De nombreuses souches bactériennes gram négatif pathogènes la produisent : Campylobacter jejuni, E. coli, Haemophilus ducreyi, Shigella dysenteriae, Salmonella typhi, etc. Cette toxine est composée de trois sous-unités (CdtA, B, C), la sous-unité active CdtB ayant des similitudes structurales et fonctionnelles avec l’ADNase I des mammifères [21]. In vitro, la protéine CDT purifiée dégrade l’ADN et la micro-injection de CDT dans un noyau induit la fragmentation de la chromatine [22]. CDT provoque des cassures double-brin dans les cellules [23] et des foyers de type IRIF sont détectés [21]. De plus, l’activité de CDT est associée à l’activation de la petite GTPase RhoA et à la formation de fibres de stress d’actine, d’une façon dépendante de la kinase ATM [23, 24]. Les cellules épithéliales ou mésenchymateuses intoxiquées avec la toxine CDT mettent souvent plusieurs jours à mourir, tandis que les lymphocytes semblent plus fragiles et meurent par apoptose [24]. Les cellules normales ou cancéreuses qui survivent à la phase aiguë d’intoxication par CDT présentent des caractéristiques de sénescence, telles que l’activation persistante des voies de réparation de l’ADN et une activité β-galactosidase typique de la sénescence [25]. CDT est la première toxine bactérienne pour laquelle on possède des informations assez précises sur le mécanisme des dommages à l’ADN. Pour le moment, une relation entre CDT et cancer n’a pas été établie.
Identification d’un nombre croissant de bactéries génotoxiques
Pseudomonas aeruginosa, un pathogène opportuniste responsable d’infections nosocomiales, est une bactérie multirésistante qui infecte principalement les individus immunodéprimés, les patients atteints de mucoviscidose, ou encore ceux subissant un geste médical invasif. La pathogénicité liée aux infections aiguës ou chroniques est multifactorielle, mais l’un des systèmes de virulence majeurs associés aux infections aiguës est le système de sécrétion de type 3 (SST3), une aiguille creuse érigée à la surface de la bactérie, au travers de laquelle les toxines bactériennes sont injectées dans le cytoplasme de la cellule hôte [26, 36]. In vitro, le SST3 est associé à des cassures double-brin de l’ADN et à des foyers nucléaires de type IRIF qui, outre γH2AX, contiennent 53BP1, la protéine de liaison à p53, impliquée dans la signalisation et la réparation des cassures [27] ( Figure 1B , étape 4). À une faible MOI, H2AX est phosphorylée dès 1 h de contact avec la bactérie et, à ce stade, il n’y a pas de signe d’apoptose ou de mort cellulaire. La toxine ExoS, via son activité ADP-ribosyltransférase, joue un rôle majeur dans l’induction de la phosphorylation d’H2AX par la kinase ATM, elle-même activée par autophosphorylation de sa sérine 1981 en présence d’ExoS [27]. ExoS n’ayant pas d’activité nucléase connue, le mécanisme directement responsable des cassures double-brin n’est pas encore identifié chez P. aeruginosa. Les cassures double-brin pourraient être consécutives à la libération de ROS et être de type oxydatif ou, comme dans le cas d’Helicobacter, être corrélées à la libération de ROS sans en être dépendantes [11].
Une autre étude sur la génotoxicité de P. aeruginosa a montré que la synthèse de OGG1 (8-oxoguanine DNA glycosylase), une enzyme de réparation de l’ADN, était activée lors d’une infection à P. aeruginosa. OGG1, un composant de la voie de réparation par excision de bases, est impliquée dans la reconnaissance et l’élimination de la 8-oxoguanine, un produit potentiellement mutagène résultant de dommages oxydatifs [28]. L’ablation du gène OGG1 (souris OGG1-/-) induit une aggravation des atteintes au niveau pulmonaire causées par une infection par P. aeruginosa, mettant en évidence le rôle important de cette protéine de réparation de l’ADN [28]. P. aeruginosa induit donc plusieurs types de cassures sur l’ADN, simple-brin ou double-brin, ce qui nécessite l’activation de plusieurs voies pour réparer les lésions. Une alternative à la mort cellulaire est envisageable si la cellule peut réparer son ADN de façon viable, notamment dans le cas d’une infection mineure associée à une antibiothérapie efficace. Cette possibilité ne constitue cependant qu’une hypothèse de travail, et aucune étude n’a abordé le sujet sous cet angle à ce jour.
La bactérie Shigella flexneri infecte les cellules épithéliales de la muqueuse intestinale du côlon, provoquant une gastroentérite aiguë, aussi appelée shigellose ou dysenterie bacillaire. La bactérie se multiplie dans les cellules infectées, provoquant une inflammation et la destruction des tissus. Elle semble utiliser un mécanisme différent de celui de E. coli et H. pylori pour parvenir à ses fins, c’est-à-dire préserver la niche épithéliale pour se répliquer alors même qu’elle induit des cassures de l’ADN [29]. La bactérie induit bien l’activation d’ATM et la phosphorylation d’H2AX, mais ces signaux n’aboutissent ni à la stabilisation de p53 ni à une mort cellulaire rapide (Figure 1B). Au contraire, la bactérie active des protéases cellulaires de la famille des calpaïnes, d’une façon dépendante du facteur de virulence VirA, ce qui engendre la dégradation de p53 [29]. La calpaïne elle-même a la propriété d’entraîner la nécrose cellulaire, mais la bactérie a le temps de proliférer dans les cellules infectées avant leur mort par nécrose [30]. Par quels mécanismes Shigella flexneri endommage-t-elle l’ADN et surtout pourquoi induit-elle une génotoxicité en premier lieu ? Ces questions restent sans réponse à l’heure actuelle.
Listeria monocytogenes, l’agent responsable de la listériose, est à l’origine de graves infections intra-utérines, de septicémies ou d’encéphalites. C’est un pathogène intracellulaire facultatif, présent sur les aliments contaminés, qui peut traverser les barrières intestinales ou placentaires, se propageant de cellule à cellule. Il a été récemment montré que la bactérie induisait des cassures double-brin de l’ADN et la production de γH2AX à de très faible MOI (moins d’une bactérie par cellule), de façon indépendante de sa toxine, la lystériolysine O (LLO) [31]. L’infection perturbe le cycle cellulaire, augmentant sa durée sans toutefois compromettre la division ou la viabilité cellulaires. En effet, des cellules infectées se divisent et donnent naissance à deux cellules filles infectées [31]. Contrairement à d’autres bactéries qui provoquent un arrêt du cycle, L. monocytogenes n’induit qu’un retard dans la phase de synthèse de l’ADN, donnant une chance à la cellule de réparer son ADN. Cette fenêtre temporelle constituerait un créneau propice à la réplication de la bactérie, qui exploite les ressources cellulaires pour se multiplier. La réponse aux dommages provoqués par L. monocytogenes semble atypique puisqu’elle induit de préférence l’activation de la kinase DNA-PK (DNA protein kinase), plutôt que celle de ses protéines apparentées ATM ou ATR (ataxia telangiectasia and Rad3-related protein) [31]. L. monocytogenes ne possède pas de toxine connue pour directement attaquer l’ADN. Elle peut inhiber la production de ROS par le biais de sa toxine LLO [32]. La bactérie mettrait en œuvre un mécanisme autre que celui des dommages oxydatifs pour casser l’ADN de la cellule hôte, mécanisme qu’il reste à élucider. Ces résultats sont à mettre en parallèle avec ceux montrant que L. monocytogenes agit sur la chromatine de la cellule hôte, en induisant la répression d’un ensemble de gènes après déacétylation de séquences régulatrices par l’histone déacétylase sirtuine 2 [33]. Il semble donc que L. monocytogenes manipule le génome eucaryote de multiples façons, bien évidemment toujours à son profit et pour favoriser sa survie.
Conclusion
Les exemples ci-dessus permettent de dégager quelques grandes idées concernant les conséquences des infections bactériennes sur le génome eucaryote. Hormis dans le cas des bactéries possédant une toxine à activité ADNase I telle que CDT, les mécanismes précis conduisant aux dommages de l’ADN (cassures double-brin, simple-brin, etc.) restent très largement inconnus à ce jour. Les infections chroniques induisent une réponse inflammatoire générant beaucoup de ROS, désignant donc les espèces réactives de l’oxygène comme les coupables idéaux. Mais est-ce la réalité ? Dans certains cas, les cassures double-brin de l’ADN se produisent dans un contexte d’inflammation chronique, un état où l’organisme est déjà fragilisé. Pour E. coli, l’inflammation chronique favoriserait l’expansion de souches plus génotoxiques que celles présentes dans le microbiote d’un organisme sain, et il existe une corrélation entre la génotoxicité et la formation de tumeurs du côlon chez la souris. Après l’induction de cassures, les bactéries agissent sur les voies de signalisation et de réparation des dommages à l’ADN. Elles manipulent le cycle cellulaire eucaryote, en l’arrêtant ou en le retardant (action sur la stabilité de p53), donnant du temps aux pathogènes intracellulaires de se répliquer avant la mort cellulaire ou la sénescence de l’hôte. Du point de vue eucaryote, la cellule infectée peut survivre dans certains cas, surtout quand les infections sont modérées. Il reste toutefois très souvent des traces de l’infection qui peuvent avoir des conséquences à court ou à long terme sur la stabilité du génome eucaryote. À l’image des rayons solaires UV, les infections répétées par des bactéries génotoxiques pourraient être une source constante de lésions de l’ADN, potentiellement dangereuses si l’effet est cumulatif dans le temps.
Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010 ; 40 : 179–204. [Google Scholar]
- Price BD, D’Andrea AD. Cell chromatin remodeling at DNA double-strand breaks. Cell 2013 ; 14 : 1344–1354. [CrossRef] [Google Scholar]
- Touati E. When bacteria become mutagenic and carcinogenic: lessons from H. pylori. Mutat Res 2010 ; 703 : 66–70. [CrossRef] [PubMed] [Google Scholar]
- Kim JJ, Tao H, Carloni E, et al. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology 2002 ; 123 : 542–553. [CrossRef] [PubMed] [Google Scholar]
- Machado AM, Figueiredo C, Touati E, et al. Helicobacter pylori infection induces genetic instability of nuclear and mitochondrial DNA in gastric cells. Clin Cancer Res 2009 ; 15 : 2995–3002. [CrossRef] [PubMed] [Google Scholar]
- Machado AM, Figueiredo C, Seruca R, Rasmussen LJ. Helicobacter pylori infection generates genetic instability in gastric cells. Biochem Biophys Acta 2010 ; 1806 : 58–65. [Google Scholar]
- Touati E, Michel V, Thiberge JM, et al. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology 2003 ; 124 : 1408–1419. [CrossRef] [PubMed] [Google Scholar]
- Umeda M, Murata-Kamiya N, Saito Y, et al. Helicobacter pylori CagA causes mitotic impairment and induces chromosomal instability. J Biol Chem 2009 ; 284 : 22166–22172. [CrossRef] [PubMed] [Google Scholar]
- Wei J, Nagy TA, Vilgelm A, et al. Regulation of p53 tumor suppressor by Helicobacter pylori in gastric epithelial cells. Gastroenterology 2010 ; 139 : 1333–1343. [CrossRef] [PubMed] [Google Scholar]
- Buti L, Spooner E, Van der Veen AG, et al. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc Natl Acad Sci USA 2011 ; 108 : 9238–9243. [CrossRef] [Google Scholar]
- Toller IM, Neelsen KJ, Steger M, et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc Natl Acad Sci USA 2011 ; 108 : 14944–14949. [CrossRef] [Google Scholar]
- El Kaoutari A, Armougom F, Raoult D, Henrissat B. Le microbiote intestinal et la digestion des polysaccharides. Med Sci (Paris) 2014 ; 30 : 259–265. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Nougayrède JP, Homburg S, Taieb F, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006 ; 313 : 848–851. [CrossRef] [PubMed] [Google Scholar]
- Cuevas-Ramos G, Petit CR, Marcq I, et al. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA 2010 ; 107 : 11537–11542. [CrossRef] [Google Scholar]
- Secher T, Samba-Louaka A, Oswald E, Nougayrède JP. Escherichia coli producing colibactin triggers premature and transmissible senescence in mammalian cells. PLoS One 2013 ; 8 : e77157. [CrossRef] [PubMed] [Google Scholar]
- Arthur JC, Perez-Chanona E, Mühlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012 ; 338 : 120–123. [CrossRef] [PubMed] [Google Scholar]
- Olier M, Marcq I, Salvador-Cartier C, et al. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes 2013 ; 3 : 501–509. [CrossRef] [Google Scholar]
- Putze J, Hennequin C, Nougayrède JP, et al. Genetic structure of the colibactin genomic island among members of the family Enterobacteriaceae. Infect Immun 2009 ; 77 : 4696–4703. [CrossRef] [PubMed] [Google Scholar]
- Lai YC, Lin AC, Chiang MK, et al. Genotoxic Klebsiella pneumoniae in Taiwan. PLoS One 2014 ; 9 : e96292. [CrossRef] [PubMed] [Google Scholar]
- Comayras C, Tasca C, Pérès SY, et al. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M. transition by preventing cdc2 protein kinase dephosphorylation and activation. Infect Immun 1997 ; 65 : 5088–5095. [PubMed] [Google Scholar]
- Lara-Tejero M, Galán JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000 ; 290 : 354–357. [CrossRef] [PubMed] [Google Scholar]
- Li L, Sharipo A, Chaves-Olarte E, et al. The Haemophilus ducreyi cytolethal distending toxin activates sensors of DNA damage and repair complexes in proliferating and non-proliferating cells. Cell Microbiol 2002 ; 4 : 87–99. [CrossRef] [PubMed] [Google Scholar]
- Frisan T, Cortes-Bratti X, Chaves-Olarte E, et al. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell Microbiol 2003 ; 5 : 695–707. [CrossRef] [PubMed] [Google Scholar]
- Guerra L, Cortes-Bratti X, Guidi R, Frisan T. The biology of the cytolethal distending toxins. Toxins (Basel) 2011 ; 3 : 172–190. [CrossRef] [PubMed] [Google Scholar]
- Blazkova H, Krejcikova K, Moudry P, et al. Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling. J Cell Mol Med 2010 ; 14 : 357–367. [CrossRef] [PubMed] [Google Scholar]
- Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 2009 ; 7 : 654–665. [CrossRef] [PubMed] [Google Scholar]
- Elsen S, Collin-Faure V, Gidrol X, Lemercier C. The opportunistic pathogen Pseudomonas aeruginosa activates the DNA double-strand break signaling and repair pathway in infected cells. Cell Mol Life Sci 2013 ; 70 : 4385–4397. [CrossRef] [PubMed] [Google Scholar]
- Wu M, Huang H, Zhang W, et al. Host DNA repair proteins in response to Pseudomonas aeruginosa in lung epithelial cells and in mice. Infect Immun 2011 ; 79 : 75–87. [CrossRef] [PubMed] [Google Scholar]
- Bergounioux J, Elisee R, Prunier AL, et al. Calpain activation by the Shigella flexneri effector VirA regulates key steps in the formation and life of the bacterium’s epithelial niche. Cell Host Microbe 2012 ; 11 : 240–252. [CrossRef] [PubMed] [Google Scholar]
- Bergounioux J, Arbibe L. Les calpaïnes, un rôle décisif dans la vie et la mort de la niche épithéliale infectée par l’entéropathogène Shigella flexneri. Med Sci (Paris) 2012 ; 28 : 1029–1071. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Leitão E, Costa AC, Brito C, et al. Listeria monocytogenes induces host DNA damage and delays the host cell cycle to promote infection. Cell Cycle 2014 ; 13 : 928–940. [CrossRef] [PubMed] [Google Scholar]
- Lam GY, Fattouh R, Muise AM, et al. Listeriolysin O suppresses phospholipase C-mediated activation of the microbicidal NADPH oxydase to promote Listeria monocytogenes infection. Cell Host Microbe 2011 ; 10 : 627–634. [CrossRef] [PubMed] [Google Scholar]
- Eskandarian HA, Impens F, Nahori MA, et al. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science 2013 ; 341 : 1238858. [CrossRef] [PubMed] [Google Scholar]
- Vielfort K, Söderholm N, Weyler L, et al. Neisseria gonorrhoeae infection causes DNA damage and affects the expression of p21, p27 and p53 in non-tumor epithelial cells. J Cell Sci 2013 ; 126 : 339–347. [CrossRef] [PubMed] [Google Scholar]
- Bischof O, Dejean A, Pineau P. Une re-vue de la sénescence. Med Sci (Paris) 2009 ; 25 : 153–160. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Lemercier C, Elsen S.. Pseudomonas aeruginosa prise en flagrant délit de casse ! Med Sci (Paris) 2013 ; 29 : 949–950. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Kaplan-Türköz B, Terradot L. Structure et mode d’injection de l’oncoprotéine CagA d’Helicobacter pylori. Med Sci (Paris) 2013 ; 29 : 33–36. [CrossRef] [EDP Sciences] [Google Scholar]
Liste des tableaux
Liste des figures
|
Figure 1. Conséquences des cassures double-brin de l’ADN et mécanismes de réparation. A. Les foyers IRIF marquant les cassures double-brin. Lors d’un stress génotoxique provoquant des cassures double-brin de l’ADN, l’histone H2AX est phosphorylée sur la sérine en position 139 (γH2AX) en quelques minutes. De nombreuses molécules γH2AX s’accumulent sur des centaines de kilobases de part et d’autres des cassures double-brin, formant des foyers nucléaires appelés IRIF (ionizing radiation-induced foci). Chaque foyer témoigne d’une cassure et est constitué d’un macro-complexe de protéines de signalisation et de réparation. En rouge γH2AX, en vert 53BP1, en bleu l’ADN. Le jaune représente la superposition des marquages rouge et vert. Échelle : 10 µm. B. Détection et signalisation des cassures double-brin de l’ADN. Une relaxation de la chromatine est initiée très rapidement au site de cassure de l’ADN (non représentée). Le senseur le plus proximal identifié est le complexe MNR, composé de Mre11, Nbs1 et Rad50, qui possède la capacité intrinsèque de lier l’ADN (étape 1). La kinase ataxia telangiectasia mutated (ATM), inactive sous forme de dimère (étape 2), est activée par autophosphorylation sur sa sérine 1981, et phosphoryle le complexe MNR, H2AX, MDC1 et p53 (étape 3). γH2AX constitue un site de liaison à haute affinité pour la protéine MDC1 qui, à son tour, orchestre le recrutement des autres effecteurs plus tardifs sur la chromatine endommagée. RNF8 et RNF168 sont deux ubiquitine-ligases qui ubiquitinent la chromatine et stimulent la fixation locale des protéines de réparation 53BP1 et BRCA1 (breast cancer 1) (étape 4). Les deux voies de réparation des cassures double-brin sont la recombinaison homologue (HR) et la voie NHEJ (non homologous end joining). La voie HR (étape 5) nécessite une matrice d’ADN dupliqué ; elle est donc active essentiellement à la phase S et G2 du cycle cellulaire. Elle assure une réparation sans erreur et sans perte d’information génétique. La voie NHEJ (étape 6) est active durant tout le cycle cellulaire et peut causer des pertes d’informations génétiques. Le cycle cellulaire est arrêté aux transitions G1/S ou G2/M, après activation de Chk1, Chk2 et Cdc25 (étape 7), donnant le temps aux mécanismes de réparation d’agir afin d’éviter la duplication du génome ou la division cellulaire alors que l’ADN est endommagé. L’apoptose dépendante de p53 intervient quand la cellule ne peut réparer son ADN (étape 8). Les points d’action des différentes bactéries citées dans le texte sont représentés. U : ubiquitine ; P : phosphorylation. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.