")
")
| Issue |
Med Sci (Paris)
Volume 30, Number 1, Janvier 2014
|
|
|---|---|---|
| Page(s) | 82 - 92 | |
| Section | Traduction | |
| Published online | 08 October 2014 | |
New hopes for metformin
Towards a better understanding of its mechanisms of action
1
Inserm U1016, Institut Cochin, département d’endocrinologie, métabolisme et diabète, 24, rue du Faubourg Saint Jacques, 75014
Paris, France
2
CNRS, UMR8104, Paris, France
3
Université Paris Descartes, Sorbonne Paris Cité, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
**
This email address is being protected from spambots. You need JavaScript enabled to view it.
Abstract
Metformin is the first-line oral antidiabetic for the treatment of type 2 diabetes. Its mechanism of action has remained unclear for many years and it is only now that the pieces of the puzzle are beginning to fall into place. Recent epidemiological data have shown that this antidiabetic drug also has a cardiovascular protective effect and some anticancer properties, aside from its action on blood glucose levels. In this article, we describe the different mechanisms of action behind the beneficial effects of metformin on blood glucose levels, cardiovascular disease and cancer.
© 2014 médecine/sciences – Inserm
The fascinating history of metformin
The history of metformin goes back to the Middle Ages in Europe and the use of the plant Galega officinalis (Figure 1), also known as French lilac or goat’s rue. This medicinal plant was used, among other things, to treat the symptoms of diabetes mellitus in humans and to increase milk production in cattle (galactogenic properties) [1]. From the 20th century onwards, the flowers and seeds of Galega were used specifically for their blood glucose-reducing (antihyperglycaemic) properties. This antihyperglycaemic effect is brought about by the active substances guanidine and isoamylene guanidine (galegine) contained in the plant (Figure 1). The latter was isolated by French pharmacist Georges Tanret in 1914. Although galegine was used with success, it was quickly abandoned because of its toxicity. In the 1920s, biguanides were produced, containing two guanidine molecules linked by an alkyl chain of varying lengths. The two biguanides synthalin A and synthalin B (decamethylene biguanidine and dodecamethylene biguanidine) (Figure 1), were used in clinical practice, but again were quickly abandoned because their therapeutic effects could not be separated from their toxic effects. Biguanides were also being synthesised at around the same time. These compounds are obtained from the condensation of two guanidine molecules and the elimination of an ammoniac molecule. Metformin (N,N-dimethylbiguanide) (Figure 1) was produced for the first time in 1922 in Dublin, by Werner and Bell. Its hypoglycaemic properties were demonstrated by two German teams in 1929 [1]. However, these discoveries were eclipsed by the discovery of insulin in 1921 and it was not until the 1950s that the clinical potential of biguanides for the treatment of diabetes was rediscovered.
 |
Figure 1. Origin, history and chemical structure of metformin and related substances. Galega officinalis was used as a medicinal plant in the Middle Ages to treat the symptoms of diabetes mellitus. In the early 20th Century, the alkaloid galegine belonging to the guanidine family was isolated as the active substance of the galega plant. Galegine has antidiabetic properties, but its toxic effects soon became apparent. Synthalin A and synthalin B, containing two guanidine molecules linked by an alkyl chain, were both used in clinical practice in the 1920s, but were also abandoned because of their toxicity. These were followed by biguanides, derived from the condensation of two guanidine molecules. Metformin was described in 1929 and tested as an antidiabetic agent in humans in 1957 by Frenchman Jean Sterne. Two other biguanides, phenformin and buformin, were used around the same time, but were withdrawn from the market as a result of their high risk of fatal acidosis. Metformin is the only drug in the biguanide class and is today the oral antidiabetic of choice for the treatment of type 2 diabetes. Photograph: Wikimedia commons, Epibase. |
In 1957, the French doctor Jean Sterne carried out the first clinical trials on metformin as an oral antidiabetic agent in humans. He demonstrated that of all the other biguanides tested, metformin had the best risk/benefit ratio [2]. Following this research, metformin was sold for the first time in France by Aron Laboratories in 1959, under the evocative brand name Glucophage. In 1958, phenformin (phenylethylbiguanide) and buformin (monobutylbiguanide) (Figure 1) were the preferred antidiabetic agents in the United States and Germany, respectively. Both of these biguanides had more potent action than metformin, but after several fatal cases of lactic acidosis and several cardiac incidents, phenformin was withdrawn from the American market in 1976. A low incidence of lactic acidosis in patients taking metformin was confirmed and the drug therefore continued to be very widely prescribed in Europe. However, the incidents linked to phenformin had tarnished the reputation of metformin by association, which prevented widespread use of the drug in the United States. The multicentre British study UKPDS (United Kingdom prospective diabetes study) confirmed metformin’s place as the treatment of choice for type 2 diabetes [3].
Having been reticent towards this ‘European’ drug in the beginning, the United States only adopted it in 1995 and it is now among the 10 most widely used medicines in the US. Metformin has been available as a generic since 2002 and is currently prescribed to over 120 million people in the world, making it the most commonly used antidiabetic drug.
Metformin’s antidiabetic effect
Metformin has been used successfully for the treatment of type 2 diabetes for over half a century1. Because of its efficacy and limited number of side effects, metformin is recommended by both the American Diabetes Association and the European Association for the Study of Diabetes as the first-line oral antidiabetic for patients with type 2 diabetes, unless contraindicated. Metformin reduces hyperglycaemia without entailing a risk of hypoglycaemia, unlike other antidiabetic agents such as sulfonylureas and insulin. This is why it is considered an antihyperglycaemic agent. Metformin also improves insulin sensitivity, leading to a reduction in insulin-resistance and lower plasma insulin levels.
The side effects associated with metformin are primarily gastrointestinal disorders, including abdominal pain, nausea, vomiting and diarrhoea. These symptoms occur at the start of treatment but usually subside quite quickly. Lactic acidosis is the most severe risk from biguanides. It is the result of increased anaerobic glycolysis, leading to blood lactate accumulation.
This in turn causes a reduced blood pH, which leads to a state of shock. Lactic acidosis was observed with phenformin but is very rare with metformin, the estimated incidence being 3 cases per 100,000 patients per year, equivalent to a risk between 10 and 20 times lower than that of phenformin. Due to the lactic acidosis risk, metformin is contraindicated in patients with any disorder that could cause either tissue hypoxia or ischaemia (such as heart or lung failure) or accumulation of metformin in the body as a result of elimination problems (for instance kidney or liver failure). However, the causal role of metformin in most reported lactic acidosis cases has not been thoroughly proven [4]. Metformin does not entail a risk of hypoglycaemia through interaction with other drugs; hence if it is not sufficient as a monotherapy it can be prescribed in combination with other antidiabetic treatments, provided there are no contraindications.
Unlike other antidiabetic treatments, metformin does not lead to weight gain; in fact obese patients are often found to lose weight [3]. This effect on weight could be a result of lower blood insulin, although an anorexigenic effect has also been suggested [5]. Metformin also has a beneficial effect on the blood lipid profile, by lowering plasma triglyceride and cholesterol concentrations, thus conferring anti-atherogenic protection [3]. Studies in both humans and mice suggest that metformin improves liver steatosis [6, 7]. Nonalcoholic fatty
liver disease is an illness often associated with type 2 diabetes, resulting from an accumulation of triglycerides in the liver. The reduction in the hepatocytes’ lipid content in response to metformin appears to limit lipotoxicity and improve insulin sensitivity in the liver [8, 9].
Furthermore, metformin helps prevent the onset of type 2 diabetes in high-risk subjects, especially those who are overweight [10], as well as improving gestational diabetes safely and effectively [11]. Polycystic ovary syndrome (PCOS) is an endocrine disorder characterised by chronic absence of ovulation associated with insulin resistance. When insulin resistance is improved using metformin in women suffering from PCOS, menstrual cycle returns and fertility is significantly improved [12].
The efficacy, safety and multiple benefits of metformin have made it the gold standard among antidiabetic treatments. Actually, strategies for drug development are based on the selection of potential drug targets. However, metformin was discovered before these drug targeting criteria became the norm and has been used for several decades without us knowing the precise molecular details of its action. For the last ten years or so, metformin has been the subject of intense research and we are now obtaining a much clearer picture of its mechanisms of action. In the following paragraphs, we will look at the latest findings on these mechanisms and potential new uses for metformin.
The liver as the target organ of metformin
The liver plays a central role in metformin’s antihyperglycaemic action. This is because the drug reduces hyperglycaemia by decreasing hepatic glucose production through inhibition of gluconeogenesis, which is abnormally high in type 2 diabetic patients. To a lesser extent, it also decreases gastrointestinal glucose absorption, while increasing glucose consumption in muscle tissue. Over time, metformin also increases insulin sensitivity in the liver.
After oral administration, metformin is absorbed by the intestine and circulating metformin concentration are high in the portal vein. As a result, the liver is exposed to higher metformin levels than other surrounding tissues. Unlike phenformin, metformin is highly hydrophilic and therefore the amount passing through the plasma membrane by passive diffusion is virtually zero. The organic cation transporters OCT1, 2 and 3 are the main transporters of metformin. OCT1 is strongly expressed in the intestine, liver and kidneys, but weakly expressed in other tissues. OCT transporters therefore participate in the absorption and elimination of metformin by the liver and kidneys, respectively. The strong expression of OCT1 in the liver leads to a higher accumulation of metformin in this organ than in other tissues. This hepatic accumulation is accentuated by the fact that metformin is not metabolised by the liver and is excreted unchanged in the urine.
These pharmacokinetic characteristics place the liver as the target organ of metformin. Deletion of the OCT 1 gene in mice leads to a strong reduction in hepatic metformin accumulation. Similarly, polymorphisms of the OCT1 gene in humans reduce the blood glucose-lowering action of metformin [13]. Metformin is also handled by the transporter MATE (multidrug and toxin extrusion), which contributes to its excretion by the kidneys, and by the transporter PMAT (plasma membrane monoamine transporter), which contributes to its intestinal absorption.
Metformin’s antihyperglycaemic action: energy is the key
The most significant discovery for our understanding of metformin’s mechanism of action at the cellular level was made by the team led by Xavier Leverve in Grenoble (France). In 1993, this group had already demonstrated that the inhibition of glucose production by metformin in hepatocytes was associated with lower intracellular ATP levels [14].
In 2000, this group showed for the first time that metformin induced a mild and specific inhibition of the mitochondrial respiratory chain complex I (NADH: ubiquinone oxidoreductase), leading to a reduction in the ATP produced by oxidative phosphorylation and a moderate increase in the AMP/ATP ratio in hepatocytes [15]. Complex I of the respiratory chain is therefore the primary target of metformin.
The precise mechanism involved in modulation of complex I activity in the respiratory chain by metformin has not been clearly established. Unlike rotenone, another complex I inhibitor, metformin is not capable of inhibiting respiration in isolated mitochondria or permeabilised cells: it is only found to have an inhibitory effect in intact cells or isolated mitochondria from cells pre-treated with metformin [15].
It is important to emphasise that this inhibition is partial and affects no more than 40% of maximum complex I activity (as opposed to 80% with rotenone), which suggests that the accumulation of metformin in the mitochondrial matrix is self-limiting. Metformin is in fact positively charged and highly hydrophilic and therefore enters the mitochondria by means of membrane potential.
The increasing concentration of metformin in the matrix gradually inhibits the respiratory chain. This will lead to a reduction in membrane potential, thus preventing it from accumulating further in the mitochondrion [16]. This self-limiting mechanism would explain the low risk of lactic acidosis found with metformin, whilst the risk is much higher with phenformin, as the latter does accumulate in the mitochondrial membrane due to its hydrophobic side chain (Figure 1). Metformin is a very effective divalent ion chelator. A recent study suggests that the effect of metformin derives from its ability to bind copper, though it does not explain the consequences within the cell [17]. Secondarily, the inhibition of complex I of the respiratory chain leads to a reduced energy state in the cell and modifies both metabolic fluxes and the activity of enzymes regulated by ATP, ADP and AMP. In particular, Zhou et al. showed in 2001 that AMPK (AMP-activated protein kinase) is activated by metformin [18]. AMPK is a phylogenetically conserved serine/threonine protein kinase [53]. This protein kinase acts as the metabolic sensor of intracellular energy and when activated, it continuously adjusts energy production and consumption inside the cell.
AMPK exist as a heterotrimer composed of a catalytic α-subunit and two regulatory β- and γ-subunits [19].
AMPK is activated allosterically by AMP and by phosphorylation on threonine-172 within the catalytic α-subunit by an AMPK kinase, identified as the tumour suppressor LKB1 (liver kinase B1). AMPK is activated by a reduction in the intracellular ATP/AMP ratio following metabolic stress such as exercise, fasting or hypoxia. Once activated, AMPK inhibits the ATP-consuming anabolic metabolic pathways and activates the ATP-producing catabolic pathways in order to restore the intracellular ATP/AMP ratio. This regulation involves the phosphorylation of key metabolic enzymes and transcription factors by AMPK, ultimately leading to modification of glucose and lipid metabolism, protein synthesis and cell growth [20].
Various molecular mechanisms have been proposed to explain the effects of metformin on gluconeogenesis in the liver, including enhancement of the action of insulin, changes in the activity of key enzymes and reduction in hepatic uptake of gluconeogenic substrates. The inhibition of gluconeogenic gene transcription by activation of the LKB1/AMPK pathway in response to metformin prevailed for several years [8, 19, 21]. However, this mechanism of action has recently been challenged by results obtained in mice lacking LKB1 or AMPK in the liver [9, 22]. These studies clearly showed that short-term inhibition of hepatic glucose production by metformin is independent of the LKB1/AMPK pathway and inhibition of gene expression.
This work also demonstrated that reduced hepatic energy state is the critical factor in inhibition of hepatic glucose production by metformin (Figure 2). This is due to the fact that gluconeogenesis is an energy-consuming metabolic pathway, given that six ATP equivalents (4 ATP and 2 GTP) are needed in order to form one glucose molecule. A reduction in intracellular ATP levels via inhibition of mitochondrial complex I therefore reduces gluconeogenic flux.
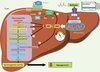 |
Figure 2. Inhibition by metformin of glucose production via reduced energy potential in the liver. Metformin is transported into hepatocytes by the transporter OCT1. Once inside the cell, it partially inhibits the mitochondrial respiratory chain at complex I, which is its primary target. This leads to a reduction in energy levels in the cell, reflected in lower intracellular ATP levels and corresponding higher AMP levels. Gluconeogensis is an energy-intensive metabolic pathway, requiring 4 ATP and 2 GTP molecules per glucose molecule produced. The reduction in ATP levels in response to metformin therefore reduces glucose production. In addition, the accumulation of AMP allosterically inhibits fructose-1,6-diphosphatase, one of the key gluconeogenesis enzymes, and reduces activation of the adenylate cyclase stimulated by glucagon. The result is slower gluconeogenesis and hence an improvement in hyperglycaemia for type 2 diabetic patients. |
In addition, increase in intracellular AMP concentrations allosterically inhibit fructose-1,6-bisphosphatase, one of the key gluconeogenesis enzymes, thus contributing to the inhibition of glucose production (Figure 2). It has recently been suggested that the increase of intracellular AMP levels induced by metformin reduces glucagon-stimulated gluconeogenesis. This is because AMP inhibits the activity of glucagon-activated adenylate cyclase by binding to an inhibitor site on the enzyme known as site P, which in turn lowers intracellular cyclic AMP concentrations and decreases protein kinase A (PKA) activity, leading to reduced glucagon-stimulated glucose production [23] (Figure 2).
Metformin therefore improves hyperglycaemia by inhibiting hepatic glucose production via a reduction in gluconeogenesis activity through falling of energy charge. This action leads to lower ATP levels and corresponding higher AMP levels, via inhibition of complex I of the respiratory chain, independently of the LKB1/AMPK pathway.
Cardioprotective effect: metformin’s secret weapon
Cardiovascular complications are still the main cause of death in patients with type 2 diabetes. In the longitudinal study UKPDS [3], it was found that taking metformin significantly reduces mortality from all causes. Unlike other antidiabetic agents (insulin and sulfonylureas), in patients taking metformin there is a 40% reduction in mortality from cardiovascular problems such as myocardial infarction and cerebrovascular incidents. This suggests that metformin’s cardiovascular protection could be separate from its antihyperglycaemic action.
Studies in non-diabetic rats has confirmed that metformin improves heart function and reduces the size of lesions after induction of myocardial infarction [24]. This beneficial effect is the result of a myocardial preconditioning induced by metformin with varying degrees of permanence. One fundamental effect of metformin is to induce metabolic adaptation of the myocardium in critical energy situations such as ischaemia. In a rodent model of heart failure, metformin promotes metabolic adaptation in the heart by stimulating gradual consumption of glucose instead of fatty acids. This action appears to be dependent upon activation of AMPK, which contributes to stimulating glucose uptake and glycolysis activity to cope with oxygen deprivation [25].
Metformin has been found to have beneficial effects in diabetic patients with a history of heart failure, reducing both mortality and morbidity [26]. One of the mechanisms thought to be behind diabetic cardiomyopathy and heart failure is defective autophagy in the heart. Metformin restores autophagy in diabetic mice and prevents the formation of cardiac lesions. AMPK must be activated, since metformin is ineffective in diabetic mice with defective AMPK activity in their hearts [27].
The cardioprotective effects of metformin stem from more than one mechanism and both macro- and microvascular systems are involved. Metformin is known to exercise a protective effect on the vascular endothelium, both by decreasing the production of free radicals and by reducing the formation of glycated proteins, which cause oxidative stress and inflammation
In view of metformin’s vascular effects and anti-atherogenic properties, its use could be extended to non-diabetic patients at risk of developing vascular disorders.
Anticancer properties: new hopes for metformin
The first studies suggesting a possible role for metformin in oncology were published in 2005 [28].
Since then, many other observational epidemiological studies have confirmed that type 2 diabetic patients who have been taking metformin for several years are at lower risk of developing or dying from cancer [29].
A relationship has also been found between the dose of metformin or duration of treatment and the protection observed, giving further evidence of anticancer properties [28]. These studies also concur on metformin’s efficacy in reducing the incidence of cancer and associated mortality across all types of cancer, although some studies have highlighted specific effects on gastrointestinal and breast cancers [29]. A wide variety of mechanisms has been described to explain its beneficial effects (Figure 3), as they are dependent upon the metabolic and molecular characteristics of each type of tumour.
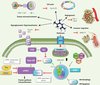 |
Figure 3. Principal mechanisms of action described for metformin’ inhibition of tumour growth. Several different modes of action have been suggested, due to the diversity of in vivo and in vitro cancer models studied. At system level, metformin reduces blood insulin and IGF1 levels, which blocks stimulation of the PI3K/AKT/mTORC1 signalling pathways and cell proliferation. At cell level, metformin inhibits the mTORC1 (mammalian target of rapamycin complex 1) pathway via mechanisms dependent on AMPK activation by phosphorylation of TSC2 (tuberous sclerosis complex 2) and raptor (regulatory associated protein of mTOR). However, metformin is able to inhibit the mTORC1 pathway via AMPK-independent mechanisms, by inhibiting Rag GTPases and inducing REDD1 expression. It is possible that the drug acts at the level of cell cycle regulation by inhibiting cyclin D1 expression. At the tumour microenvironment level, stimulation of memory T cell generation also contributes to plays a part in metformin’s beneficial effects. Metformin causes a reduction in neoplastic cell angiogenesis by lowering blood concentrations of PAI-1 (plasminogen activator inhibitor-1) and VEGF (vascular endothelial growth factor). Inhibition of metastasis formation through decreased activity of the metalloproteinases MMP2 and -9 has also been described. |
Effect of metformin on blood insulin and IGF1 levels
Several epidemiological studies have reported a strong link between type 2 diabetes and elevated risk of certain types of cancer, in particular colon, endometrium, rectum and breast cancer, compared with the incidence of these cancers in the non-diabetic population. It is thought that compensatory hyperinsulinaemia and chronic hyperglycaemia, both characteristics of type 2 diabetes, are behind this link between diabetes and cancer. Insulin is known to promote cell proliferation and increase blood levels of IGF1 (insulin-like growth factor 1), which acts as a growth factor in tumour development. It is also well established that glucose is the preferred energy substrate used by cells during proliferation and hyperglycaemia could therefore contribute to increasing the growth and survival of cancer cells.
Certain treatments for reducing hyperglycaemia and/or hyperinsulinaemia have been found to reduce the risk of cancer. The inhibition of tumour progression by metformin has been confirmed through studies of large numbers of mouse carcinogenesis models, as well as the observational epidemiological studies. In some models, the inhibition of tumour proliferation was directly linked to reduction in blood insulin and IGF1 levels by metformin (Figure 3) and reduced activity of receptors with tyrosine kinase activity [30]. However, this effect was not observed in other models, suggesting a mode of action working independently of decreased blood insulin levels.
Metformin’s antiproliferative action
Several clinical trials have suggested that metformin is able to procure a protective effect against cancer regardless of whether the patient has diabetes [31]. This finding has been supported by a large body of in vitro evidence demonstrating that the drug directly inhibits the proliferation of cancer cells [32, 33]. It is important to point out that tumour cells have varying degrees of sensitivity to metformin and its antiproliferative effect is only observed at concentrations much higher (5-50 mM) than those used to treat diabetes (plasma concentrations in the region of 10–40 μM). Nonetheless, metformin does accumulate in some organs, such as the intestine and liver, allowing much higher intracellular concentrations than those in the plasma.
Regulation of the AMPK/ mTOR pathway
Several studies have suggested that metformin’s antiproliferative effects could be dependent upon activation of AMPK following inhibition of complex I of the respiratory chain. To support this assumption, inhibition of AMPK or the use of LKB1-deficient cells blocks the drug’s antiproliferative effect [32]. Moreover, in a carcinogenesis model induced via the loss of the tumour suppressor PTEN (phosphatase and tensin homolog), activation of AMPK by a direct activator (A-769662) produces a reduction in tumour progression identical to that obtained with metformin [34]. AMPK inhibits the mTORC1 (mammalian target of rapamycin complex 1) signalling pathway, which integrates nutrient and mitogen signals via the Ras/ERK (extracellular signal-regulated kinase) and PI3K (phosphoinositide 3-kinase)/AKT (protein kinase B) pathways, thus controlling cell growth and proliferation [54].
AMPK directly phosphorylates the tumour suppressor TSC2 (tuberous sclerosis complex 2) and the regulator protein raptor (regulatory associated protein of mTOR), leading to rapid suppression of mTORC1 signalling pathway (Figure 3). Although the LKB1/AMPK pathway is described as a key component in metformin’s inhibition of mTORC1 pathway, other, AMPK-independent mechanisms involving Rag GTPases or REDD1 (regulated in development and DNA damage response 1) have been suggested [35] (Figure 3). This inhibition of the mTORC1 pathway by metformin leads to decreased expression of EGF (epidermal growth factor) receptor and the oncoprotein HER2 (erbB-2) in breast cancer lines.
Cell cycle regulation
A study of gene expression profiles in breast cancer tumours treated with metformin revealed a reduction in the expression of genes involved in mitosis progression. Similarly, treatment of lung and breast cancer cells with metformin interrupts the cell cycle at the G2/M phase and increases apoptosis. In prostate cancer cell lines, metformin induces cell cycle arrest in the G0/G1 phase via reduced expression of cyclin D1 [33] (Figure 3).
Metabolic reprogramming of cancer cells
The metabolism of cancer cells is essentially based on consuming glucose to produce energy, even when oxygen is present. This feature means that cancer cells preferentially use glycolysis in aerobic conditions (Warburg effect [52] or aerobic glycolysis), as opposed to mitochondrial oxidative phosphorylation. However, cancer cells use their mitochondria to produce some metabolites (malate, citrate) needed for the various biosynthetic pathways required for cell proliferation.
By blocking the respiratory chain, metformin will therefore inhibit this adaptive metabolism and cause a major energy crisis. This will force the cancer cells into a survival process (inducing glycolysis and/or autophagy), which will fail and lead to cell death by apoptosis. For example, tumour cells with a loss of p53 function cannot cope with the metabolic changes imposed by metformin (which require activation of p53 by AMPK) and die from apoptosis [36]. Similarly, cancer cells that are deficient in LKB1 (showing low AMPK activity) are more sensitive to the ATP depletion imposed by metformin because of their inability to restore their energy balance via AMPK activation. In these situations, metformin acts on cancer cells as a selective cytotoxic agent.
Metformin’s action on the tumour microenvironment
Changes in the tumour microenvironment may also contribute to the beneficial effects of metformin. The drug has been reported to stimulate the generation of CD8+ memory T cells, thus inducing greater anticancer immunity [37]. The possibility has also been raised that it may inhibit tumour angiogenesis via a reduction in blood levels of PAI-1 (plasminogen activator inhibitor-1) and VEGF (vascular endothelial growth factor) (Figure 3). However, the opposite was found in a different study, reporting an increase in intratumoural microvascular density in tumour xenograft cells [31].
Metformin’s preventive action on tumour development
Metformin participates in the response to damaged DNA by selectively activating ATM (ataxia telangiectasia mutated), but also by inhibiting reactive oxygen species production brought about through transformation by the oncogene Ras [38]. Metformin also contributes to preventing tumour development by controlling cancer stem cell ontogenesis, as it targets the epithelial-mesenchymal transition and differentiation in these cells [39, 40]. This specific action of metformin on cancer stem cells has opened up new possibilities in cancer prevention, due to the resistance of these cells to chemotherapy.
It is significant that metformin treatment has been linked to a better response rate to chemotherapy for breast cancer [41]. This synergistic effect of metformin on the cytotoxicity of chemotherapy agents has been confirmed in vitro and has been found to be effective in reducing resistance to these drugs, which supports the potential use of metformin as a chemotherapy adjuvant [42]. Lastly, recent studies have suggested a role for metformin in inhibiting the invasive potential of cancer cells and the formation of metastasis by decreasing the activity of the metalloproteinases MMP2 and MMP9 [40, 43] (Figure 3).
So, after more than 50 years’ service as a diabetes treatment, this old drug may find a new lease of life as prospects open up for its use in cancer. However, it is now important to identify which patients might benefit from its potential for preventing and treating cancer, by investigating biomarkers that will predict its therapeutic effect. The sensitivity of cancer cells to metformin depends on the type of cancer, the presence of mutations (for example for LKB1, p53 or OCT1 polymorphisms) and the tumour environment.
On the basis of the pharmacokinetic properties of phenformin (membrane permeability, potent complex I inhibition), several authors have suggested that this drug previously taken off the market could be used as a much more effective antineoplastic agent than metformin [34]. Phenformin has extremely potent anticancer properties under metabolic stress conditions when the LKB1/AMPK pathway is not operational [44]. Future studies will need to determine the optimal tolerable doses of both metformin and phenformin for the treatment of different cancers. Over one hundred phase II and III clinical trials are currently under way to assess the use of metformin in oncology2.
Neuroprotective properties of metformin: hope for neurogenerative diseases?
Recent studies have opened up the prospect of possible therapeutic indications for metformin in the treatment of neurodegenerative diseases. Metformin may have a neuroprotective and prophylactic effect in patients predisposed to Alzheimer’s disease [45]. Hyperphosphorylation of microtubule-associated protein tau causes neurofibrillary tangles and is a major factor in Alzheimer’s pathogenesis. Metformin induces dephosphorylation of tau by activating the protein phosphatase 2A (PP2A), and thus slows the progression of the disease.
Other targets of metformin’s preservative action on the central nervous system are inhibition of inflammation and oxidative stress, as demonstrated in an experimental autoimmune encephalomyelitis murine model of multiple sclerosis [46]. A potential effect of metformin on cognitive function has been suggested, after it was found to delay the onset of cognitive disorders in a mouse model of Huntington’s disease [47]. Another recent study also showed that metformin promotes neurogenesis and enhances hippocampus-dependent spatial memory formation and learning [48]. Metformin promotes phosphorylation of CBP (CREB binding protein), a transcription coactivator with intrinsic histone acetyltransferase activity, via the protein kinase C (PKC)-ζ. This stage is crucial for the differentiation of radial glial neuronal precursors and the formation of new neurons in the hippocampus.
Effect on longevity: metformin as an elixir of life?
The protection afforded by metformin against diabetes, cardiovascular disease and cancer is similar to the anti-ageing effects of calorie restriction (Figure 4). Dietary restriction by 25–60% is a well-known means of increasing life expectancy in all animal species. There have been reports of metformin having properties that mimic calorie restriction and prolong the lifespan of the nematode Caenorhabditis elegans and rodents.
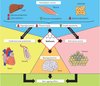 |
Figure 4. Summary of the principal effects of metformin. The principal antidiabetic effects of metformin occur in the liver, via inhibition of gluconeogenesis, and to a lesser extent in the intestine and muscle, leading to a reduction in hyperglycaemia and blood lipids and to increased insulin sensitivity. These same improvements indirectly procure both cardiovascular protective and anticancer effects. Added to this, metformin directly reduces the risk of cardiovascular disease through actions that affect both macro- and microvascular systems and exerts a direct antitumoural effect on cancer cells. Furthermore, metformin is thought to have a neuroprotective effect in neurodegenerative diseases. The beneficial effects of metformin are similar to those observed with calorie restriction, which help to prolong lifespan. Metformin can therefore be considered a calorie restriction mimetic with possible anti-ageing properties. |
In non-diabetic mice, metformin mimics the effects of calorie restriction on the gene expression profile and prevents the onset of diabetes, cardiovascular disease and cancer [49]. Metformin’s effects in nematodes are dependent upon the LKB1/AMPK pathway and involve antioxidant defense longevity pathways [50]. It has recently been suggested that metformin indirectly increases the lifespan of nematodes by disrupting the metabolism of its accompanying microbe Escherichia coli [51]. This raises the question as to whether similar mechanisms may exist in humans, altering the metabolism of gut microflora, and whether these alterations may contribute to metformin’s therapeutic effects.
Conclusion
At present, metformin is the most frequently used antidiabetic agent in the treatment of type 2 diabetes. Its success is due to various factors: efficacy, safety, good tolerability and low production cost. The liver is the main target of metformin’s action, as it moderately inhibits complex I of the mitochondrial respiratory chain. This causes a reduction in the liver’s energy state, which in turn leads to a decrease in hepatic glucose production.
Under these energy stress conditions, activation of the energy sensor AMPK does not, however, play a role in metformin’s gluconeogenesis inhibition. The drug also has the advantage of counteracting the cardiovascular complications associated with diabetes, by inducing myocardial preconditioning. Another significant benefit of metformin is its demonstrated capacity to reduce the risk of tumour development by controlling cell differentiation and proliferation. New therapeutic indications may also be found for the drug in the treatment of neurodegenerative diseases. This old drug may well have more surprises in store.
Declaration of interests
The authors declare that they have no interests associated with the information published in this article.
See ‘Diabetes’ special issue of médecine/sciences, August-September 2013.
The list may be viewed at http://www.clinicaltrials.gov
References
- Bailey C, Campbell I. Metformin: the gold standard; a scientific handbook Chichester, UK: Wiley, 2007: 288 p. [Google Scholar]
- Sterne J. Du nouveau dans les antidiabétiques. La NN diméthylamino guanyl guanidine (N.N.D.G.). Maroc Med 1957; 36: 1295–1296. [Google Scholar]
- UKPDS. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998; 352: 854–865. [CrossRef] [PubMed] [Google Scholar]
- Stades AM, Heikens JT, Erkelens DW, et al. Metformin and lactic acidosis: cause or coincidence? A review of case reports. J Intern Med 2004; 255: 179–187. [CrossRef] [PubMed] [Google Scholar]
- Lee A, Morley JE. Metformin decreases food consumption and induces weight loss in subjects with obesity with type II non-insulin-dependent diabetes. Obes Res 1998; 6: 47–53. [CrossRef] [PubMed] [Google Scholar]
- Lin HZ, Yang SQ, Chuckaree C, et al. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med 2000; 6: 998–1003. [CrossRef] [PubMed] [Google Scholar]
- Marchesini G, Brizi M, Bianchi G, et al. Metformin in non-alcoholic steatohepatitis. Lancet 2001; 358: 893–894. [CrossRef] [PubMed] [Google Scholar]
- Foretz M, Viollet B. Mécanisme d’action hépatique de la metformine dans le diabète de type 2. Med Mal Metab 2009; 3: 48–54. [Google Scholar]
- Foretz M, Viollet B. Mécanisme d’inhibition de la production hépatique de glucose par la metformine. Med Sci (Paris) 2010; 26: 663–666. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346: 393–403. [CrossRef] [PubMed] [Google Scholar]
- Rowan JA, Hague WM, Gao W, et al. Metformin versus insulin for the treatment of gestational diabetes. N Engl J Med 2008; 358: 2003–2015. [CrossRef] [PubMed] [Google Scholar]
- Lord JM, Flight IH, Norman RJ. Metformin in polycystic ovary syndrome: systematic review and meta-analysis. BMJ 2003; 327: 951–953. [CrossRef] [PubMed] [Google Scholar]
- Shu Y, Sheardown SA, Brown C, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest 2007; 117: 1422–1431. [CrossRef] [PubMed] [Google Scholar]
- Argaud D, Roth H, Wiernsperger N, Leverve XM. Metformin decreases gluconeogenesis by enhancing the pyruvate kinase flux in isolated rat hepatocytes. Eur J Biochem 1993; 213: 1341–1348. [CrossRef] [PubMed] [Google Scholar]
- El-Mir MY, Nogueira V, Fontaine E, et al. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 2000; 275: 223–228. [CrossRef] [PubMed] [Google Scholar]
- Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its antidiabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 2000; 348: 607–614. [CrossRef] [PubMed] [Google Scholar]
- Logie L, Harthill J, Patel K, et al. Cellular responses to the metal-binding properties of metformin. Diabetes 2012; 61: 1423–1433. [CrossRef] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001; 108: 1167–1174 Foretz M, Taleux N, Guigas B, et al. Régulation du métabolisme énergétique par l’AMPK. Med Sci (Paris) 2006; 22: 381-8.. [CrossRef] [PubMed] [Google Scholar]
- Foretz M, Viollet B. Regulation of hepatic metabolism by AMPK. J Hepatol 2011; 54: 827–829. [CrossRef] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005; 310: 1642–1646. [CrossRef] [PubMed] [Google Scholar]
- Foretz M, Hebrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest 2010; 120: 2355–2369. [CrossRef] [PubMed] [Google Scholar]
- Miller RA, Chu Q, Xie J, et al. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013; 494: 256–260. [CrossRef] [PubMed] [Google Scholar]
- Yin M, van der Horst IC, van Melle JP, et al. Metformin improves cardiac function in a nondiabetic rat model of post-MI heart failure. Am J Physiol Heart Circ Physiol 2011; 301: H459–H468. [CrossRef] [PubMed] [Google Scholar]
- Gundewar S, Calvert JW, Jha S, et al. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res 2009; 104: 403–411. [CrossRef] [PubMed] [Google Scholar]
- MacDonald MR, Eurich DT, Majumdar SR, et al. Treatment of type 2 diabetes and outcomes in patients with heart failure: a nested case-control study from the U.K. General practice research database. Diabetes care 2010; 33: 1213–1218. [CrossRef] [PubMed] [Google Scholar]
- Xie Z, Lau K, Eby B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 2011; 60: 1770–1778. [CrossRef] [PubMed] [Google Scholar]
- Evans JM, Donnelly LA, Emslie-Smith AM, et al. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005; 330 : 1304–1305. [CrossRef] [PubMed] [Google Scholar]
- Beck E, Scheen AJ. Quels bénéfices antitumoraux attendre de la metformine ? Ann Endocrinol (Paris) 2013; 74: 137–147. [CrossRef] [PubMed] [Google Scholar]
- Memmott RM, Mercado JR, Maier CR, et al. Metformin prevents tobacco carcinogen-induced lung tumorigenesis. Cancer Prev Res 2010; 3 : 1066–1076. [CrossRef] [PubMed] [Google Scholar]
- Viollet B, Foretz M. Metformine et cancer. Du diabète au cancer: de nouvelles perspectives thérapeutiques pour la metformine. Med Mal Metab 2011; 5: 29–37. [Google Scholar]
- Zakikhani M, Dowling R, Fantus IG, et al. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 2006; 66 : 10269–10273. [CrossRef] [PubMed] [Google Scholar]
- Ben Sahra I, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008; 27: 3576–3586. [CrossRef] [PubMed] [Google Scholar]
- Huang X, Wullschleger S, Shpiro N, et al. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J 2008; 412: 211–221. [CrossRef] [PubMed] [Google Scholar]
- Ben Sahra I, Regazzetti C, Robert G, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res 2011; 71: 4366–4372. [CrossRef] [PubMed] [Google Scholar]
- Buzzai M, Jones RG, Amaravadi RK, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 2007; 67: 6745–6752. [CrossRef] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009; 460: 103–107. [CrossRef] [PubMed] [Google Scholar]
- Algire C, Moiseeva O, Deschenes-Simard X, et al. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev Res 2012; 5: 536–543. [CrossRef] [Google Scholar]
- Del Barco S, Vazquez-Martin A, Cufi S, et al. Metformin: multi-faceted protection against cancer. Oncotarget 2011; 2: 896–917. [PubMed] [Google Scholar]
- Cerezo M, Tichet M, Abbe P, et al. Metformin blocks melanoma invasion and metastasis development in a AMPK/p53-dependent manner. Mol Cancer Ther 2013; 12: 1605–1615. [CrossRef] [PubMed] [Google Scholar]
- Jiralerspong S, Palla SL, Giordano SH, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol 2009; 27: 3297–3302. [CrossRef] [PubMed] [Google Scholar]
- Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res 2009; 69: 7507–7511. [CrossRef] [PubMed] [Google Scholar]
- Hwang YP, Jeong HG. Metformin blocks migration and invasion of tumour cells by inhibition of matrix metalloproteinase-9 activation through a calcium and protein kinase Calpha-dependent pathway: phorbol-12- myristate-13-acetate-induced/extracellular signal-regulated kinase/ activator protein-1. Br J Pharmacol 2010; 160: 1195–1211. [CrossRef] [PubMed] [Google Scholar]
- Shackelford DB, Abt E, Gerken L, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013; 23: 143–158. [CrossRef] [PubMed] [Google Scholar]
- Kickstein E, Krauss S, Thornhill P, et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci USA 2010; 107 : 21830–21835. [CrossRef] [Google Scholar]
- Paintlia AS, Paintlia MK, Mohan S, et al. AMP-activated protein kinase signaling protects oligodendrocytes that restore central nervous system functions in an experimental autoimmune encephalomyelitis model. Am J Pathol 2013; 183: 526–541. [CrossRef] [PubMed] [Google Scholar]
- Ma TC, Buescher JL, Oatis B, et al. Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci Lett 2007; 411: 98–103. [CrossRef] [PubMed] [Google Scholar]
- Wang J, Gallagher D, DeVito LM, et al. Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell 2012; 11: 23–35. [CrossRef] [PubMed] [Google Scholar]
- Anisimov VN, Berstein LM, Popovich IG, et al. If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging (Albany NY) 2011; 3: 148–157. [PubMed] [Google Scholar]
- Onken B, Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS One 2010; 5: e8758. [CrossRef] [PubMed] [Google Scholar]
- Cabreiro F, Au C, Leung KY, et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 2013; 153: 228–239. [CrossRef] [PubMed] [Google Scholar]
- Razungles J, Jalaguier S, Cavaillès V, Teyssier C. L’effet Warburg: de la théorie du cancer aux applications thérapeutiques en cancérologie. Med Sci (Paris) 2013; 11: in press. [Google Scholar]
- Foretz M, Taleux N, Guigas B, et al. Regulation of energy metabolism by AMPK: a novel therapeutic approach for the treatment of metabolic and cardiovascular diseases. Med Sci (Paris) 2006; 22: 381–388. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Julien LA, Roux PP. mTOR, the mammalian target of rapamycin. Med Sci (Paris) 2010; 26: 1056–1060. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
All Figures
|
Figure 1. Origin, history and chemical structure of metformin and related substances. Galega officinalis was used as a medicinal plant in the Middle Ages to treat the symptoms of diabetes mellitus. In the early 20th Century, the alkaloid galegine belonging to the guanidine family was isolated as the active substance of the galega plant. Galegine has antidiabetic properties, but its toxic effects soon became apparent. Synthalin A and synthalin B, containing two guanidine molecules linked by an alkyl chain, were both used in clinical practice in the 1920s, but were also abandoned because of their toxicity. These were followed by biguanides, derived from the condensation of two guanidine molecules. Metformin was described in 1929 and tested as an antidiabetic agent in humans in 1957 by Frenchman Jean Sterne. Two other biguanides, phenformin and buformin, were used around the same time, but were withdrawn from the market as a result of their high risk of fatal acidosis. Metformin is the only drug in the biguanide class and is today the oral antidiabetic of choice for the treatment of type 2 diabetes. Photograph: Wikimedia commons, Epibase. |
| In the text | |
|
Figure 2. Inhibition by metformin of glucose production via reduced energy potential in the liver. Metformin is transported into hepatocytes by the transporter OCT1. Once inside the cell, it partially inhibits the mitochondrial respiratory chain at complex I, which is its primary target. This leads to a reduction in energy levels in the cell, reflected in lower intracellular ATP levels and corresponding higher AMP levels. Gluconeogensis is an energy-intensive metabolic pathway, requiring 4 ATP and 2 GTP molecules per glucose molecule produced. The reduction in ATP levels in response to metformin therefore reduces glucose production. In addition, the accumulation of AMP allosterically inhibits fructose-1,6-diphosphatase, one of the key gluconeogenesis enzymes, and reduces activation of the adenylate cyclase stimulated by glucagon. The result is slower gluconeogenesis and hence an improvement in hyperglycaemia for type 2 diabetic patients. |
| In the text | |
|
Figure 3. Principal mechanisms of action described for metformin’ inhibition of tumour growth. Several different modes of action have been suggested, due to the diversity of in vivo and in vitro cancer models studied. At system level, metformin reduces blood insulin and IGF1 levels, which blocks stimulation of the PI3K/AKT/mTORC1 signalling pathways and cell proliferation. At cell level, metformin inhibits the mTORC1 (mammalian target of rapamycin complex 1) pathway via mechanisms dependent on AMPK activation by phosphorylation of TSC2 (tuberous sclerosis complex 2) and raptor (regulatory associated protein of mTOR). However, metformin is able to inhibit the mTORC1 pathway via AMPK-independent mechanisms, by inhibiting Rag GTPases and inducing REDD1 expression. It is possible that the drug acts at the level of cell cycle regulation by inhibiting cyclin D1 expression. At the tumour microenvironment level, stimulation of memory T cell generation also contributes to plays a part in metformin’s beneficial effects. Metformin causes a reduction in neoplastic cell angiogenesis by lowering blood concentrations of PAI-1 (plasminogen activator inhibitor-1) and VEGF (vascular endothelial growth factor). Inhibition of metastasis formation through decreased activity of the metalloproteinases MMP2 and -9 has also been described. |
| In the text | |
|
Figure 4. Summary of the principal effects of metformin. The principal antidiabetic effects of metformin occur in the liver, via inhibition of gluconeogenesis, and to a lesser extent in the intestine and muscle, leading to a reduction in hyperglycaemia and blood lipids and to increased insulin sensitivity. These same improvements indirectly procure both cardiovascular protective and anticancer effects. Added to this, metformin directly reduces the risk of cardiovascular disease through actions that affect both macro- and microvascular systems and exerts a direct antitumoural effect on cancer cells. Furthermore, metformin is thought to have a neuroprotective effect in neurodegenerative diseases. The beneficial effects of metformin are similar to those observed with calorie restriction, which help to prolong lifespan. Metformin can therefore be considered a calorie restriction mimetic with possible anti-ageing properties. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.