")
")
| Issue |
Med Sci (Paris)
Volume 27, Number 10, Octobre 2011
|
|
|---|---|---|
| Page(s) | 833 - 841 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20112710011 | |
| Published online | 21 October 2011 | |
Immunothérapie des cancers
Succès récents et perspectives
Cancer immunotherapy: recent breakthroughs and perspectives
1
Université Paris Descartes, Sorbonne Paris Cité, Inserm U970, PARCC ; AP-HP, Hôpital européen Georges Pompidou, Service d’immunologie biologique, 20, rue Leblanc, 75015
Paris, France
2
Université Paris Descartes, Sorbonne Paris Cité, Inserm U970, PARCC (Paris-Centre de recherche cardiovasculaire), 56, rue Leblanc, 75015 Paris, France
3
Transgene SA, Parc d’innovation, 67405 Illkirch, France
4
AP-HP, Hôpital européen Georges Pompidou, Service d’immunologie biologique ; Inserm, unité mixte de recherche (UMR) S872, Centre de recherche des Cordeliers, Université Pierre et Marie Curie-Paris 6 et Université Paris Descartes, Sorbonne Paris Cité, 15, rue de l’École de Médecine, 75005
Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
L’immunothérapie des cancers a longtemps été considérée comme une approche thérapeutique séduisante mais peu efficace. Deux nouvelles approches d’immunothérapie, l’une vaccinale chez des patients atteints de cancers de la prostate ou de mélanomes, et l’autre utilisant un anticorps anti-CTLA4 qui cible les lymphocytes T de patients atteints de mélanomes, ont démontré leur efficacité dans des essais cliniques de phase III. La prise en compte de l’immunosuppression associée au cancer et la combinaison de l’immunothérapie avec les traitements conventionnels (chimiothérapie, traitement antiangiogénique) devraient encore améliorer cette approche, aussi bien dans son efficacité que dans ses indications cliniques.
Abstract
Immunotherapy of cancer has long been considered as an attractive therapeutic approach but with no impact on clinical practice. Two clinical protocols of immunotherapy, one based on a cancer vaccine in patients with prostate cancer or melanoma and the other using an immunomodulator targeting T cells (anti-CTLA4 mAb) in melanoma patients, have demonstrated clinical efficacy in two phase III clinical trials. To improve these encouraging clinical results, biomarkers to better select patients which may benefit from this therapy are actively searched. In addition, immunosuppression associated with cancer has to be overcome to allow a better immunostimulation. In contrast to chemotherapy, clinical variables to monitor the efficacy of immunotherapy has to be revisited and overall survival appears to be a better endpoint than clinical response defined by the RECIST criteria. Combination of immunotherapy with conventional treatments (chemotherapy, anti-angiogenic, etc.) should further improve this approach both in its effectiveness and in its clinical indications.
© 2011 médecine/sciences – Inserm / SRMS
L’histoire de l’immunothérapie est riche d’espoirs le plus souvent déçus. En dehors du BCG dans les cancers de la vessie, de l’interleukine (IL)-2 dans les tumeurs du rein et de l’interféron (IFN)-α de façon très ponctuelle dans quelques tumeurs, la place de l’immunothérapie en cancérologie restait très marginale [1].
Au cours de ces dernières années, cette vision négative s’est d’abord atténuée grâce au succès des anticorps thérapeutiques en cancérologie, qui a constitué une avancée thérapeutique majeure. Ainsi, des anticorps contre Her2/neu (trastuzumab : Herceptine®), CD20 (rituximab : Mabthéra®), le récepteur de l’EGF (cétuximab : Erbitux®) ou le VEGF (vascular endothelial growth factor) (bévacizumab : Avastin®) ont démontré leur efficacité et sont prescrits dans un nombre de plus en plus important de tumeurs (cancers du sein, lymphomes, tumeurs du côlon, cancers du rein, tumeurs de la sphère otorhinolaryngée, cancers du poumon). Ces anticorps, qui agissent directement sur la cellule tumorale ou via l’inhibition de molécules proangiogéniques, ont été perçus comme étant de nouvelles chimiothérapies moins toxiques, mais n’ont pas été considérés comme une approche d’immunothérapie active. En fait, il est aujourd’hui admis que leurs mécanismes d’action mettent en jeu le recrutement d’effecteurs immunologiques, et qu’une partie de leur action cytotoxique est médiée par des cellules (macrophages, cellules natural killer) liant l’anticorps par son fragment Fc, ou via l’activation de la voie classique du complément [56] (Figure 1).
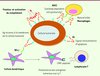 |
Figure 1. Mode d’action des anticorps. Certains anticorps (Ac) peuvent bloquer la liaison de ligands sur des récepteurs de facteurs de croissance ou provoquer un signal direct d’apoptose vers la cellule tumorale (exemple : anticorps anti-EGFR). La majorité des anticorps vont recruter des cellules effectrices (NK, macrophages) qui vont entraîner la lyse de la cellule tumorale par un mécanisme d’ADCC (antibody dependent cell cytotoxicity). La partie Fab (variable) de l’anticorps se fixe sur la cellule tumorale et la partie Fc de l’anticorps se fixe sur des récepteurs Fcγ activateurs (RFcγI, IIA, IIIA) à la surface des cellules effectrices. Ce pontage entre la cellule effectrice et la cellule tumorale par l’anticorps déclenche la cytotoxicité médiée par les cellules exprimant les RFc activateurs. Des cellules dendritiques peuvent également internaliser la cellule tumorale et présenter des peptides dérivés d’antigènes tumoraux aux lymphocytes T. |
Plus récemment, chez la souris, l’efficacité clinique de certains anticorps thérapeutiques a aussi été reliée à leur capacité à induire des lymphocytes T antitumoraux en favorisant la présentation de l’antigène tumoral aux lymphocytes T [2]. Chez l’homme, des lymphocytes T CD4+ et une réponse anticorps endogène ont été détectés après administration de trastuzumab [3]. Les réponses immunitaires cellulaire et humorale endogènes induites par l’administration d’anticorps sont le plus souvent dirigées contre la même cible que celle de l’anticorps thérapeutique. Ainsi la réponse lymphocytaire T mémoire générée pourrait être un élément-clé de la durabilité de la réponse aux anticorps monoclonaux. Ces traitements par anticorps monoclonaux, qui ont fait l’objet d’un numéro thématique complet de médecine/sciences en décembre 2009 [4], ne doivent donc plus être considérés seulement comme une immunothérapie passive.
Si l’efficacité de ces anticorps monoclonaux ravivait quelques lueurs d’espoir, la majorité des immunothérapeutes depuis plus de vingt ans cherchent à guérir le cancer en activant les lymphocytes T antitumoraux. Cette approche se justifie par l’observation bien documentée d’une réponse naturelle lymphocytaire T reconnaissant différents types d’antigènes tumoraux bien caractérisés sur le plan moléculaire (Tableau I). La mise en évidence de lymphocytes T antitumoraux dans d’exceptionnelles observations de régressions spontanées de tumeurs chez l’homme, et le bon pronostic généralement associé aux tumeurs où existe une infiltration par des lymphocytes T renforçaient cette hypothèse de travail [5]. Le groupe de Rosenberg a validé ce rôle positif des lymphocytes T lors de nombreux protocoles d’immunothérapie adoptive où des lymphocytes T autologues de patients, activés et amplifiés in vitro, étaient réadministrés in vivo [6]. Les taux de réponse variaient de 20 % à 50 %. Néanmoins, même si des techniques plus récentes facilitent cette approche, elle reste limitée à quelques centres de référence et n’est pas généralisable. Ces limites expliquent le développement depuis plus de 15 ans de nombreux protocoles de vaccination antitumorale ou d’activation des lymphocytes T directement in vivo. Aucune démonstration d’un bénéfice thérapeutique pour le patient n’avait été apportée jusqu’au récent succès de deux protocoles d’immunothérapie de phase III.
Classification et quelques exemples d’antigènes tumoraux chez l’homme. Les antigènes (Ag) de différenciation sont spécifiques d’un tissu donné. Ils seront exprimés par les cellules normales et les cellules tumorales dérivées de ces tissus. Les antigènes du groupe « cancer testis » sont exprimés par les cellules tumorales et les cellules germinales [59]. L’absence d’expression de molécules HLA par les cellules germinales limite le risque d’auto-immunité en cas de réponse antitumorale ciblant ces antigènes. Le fait que 10 % des cancers soient associés à des virus peut permettre d’envisager une vaccination prophylactique contre certains cancers (cancers du foie, du col de l’utérus, etc.). Abréviations : voir Glossaire.
Premiers succès d’essais cliniques d’immunothérapie chez l’homme
Sipuleucel-T : premier vaccin thérapeutique commercialisé chez l’homme
En juin 2010, la FDA (Food and drug administration aux États-Unis), après trois essais de phase III, a approuvé la commercialisation du sipuleucel-T, premier vaccin thérapeutique destiné à des patients atteints de cancer de la prostate résistant aux antiandrogènes mais sans métastases viscérales [7]. Le vaccin cellulaire comprenait les cellules mononucléées autologues du patient sensibilisées in vitro avec une protéine de fusion associant la phosphatase acide prostatique (PAP) couplée au GM-CSF (granulocyte-macrophage colony-stimulating factor). Le GM-CSF permet de recruter des cellules myéloïdes et peut-être d’engager leur différenciation en cellules dendritiques. Deux groupes de patients étaient vaccinés, soit avec le vaccin cellulaire soit avec les cellules mononucléées sans l’antigène. Les concentrations sériques en PSA (prostate specific antigen) et LDH (lactate deshydrogénases) avant traitement étaient similaires dans les deux groupes. La diminution du risque de mortalité était significative : ainsi, 36 mois après le début du traitement, 31,7 % des patients ayant reçu le vaccin étaient vivants mais seulement 23 % dans le groupe ayant reçu le placebo, ce qui représente une amélioration de la survie globale de 4,1 mois (25,8 mois versus 21,7 mois, p = 0,03). Pour ce type de patients résistants à la chimiothérapie, ce résultat est significatif. Une prolifération de lymphocytes T spécifiques de la protéine PAP-GM-CSF était observée chez 73 % des patients et une production d’anticorps (réponse humorale) chez 66,2 %. La présence d’anticorps et la survie étaient corrélées. Le vaccin n’avait pas d’impact sur la survie sans progression1 et n’avait pas d’effets mesurés sur les critères RECIST (response evaluation criteria in solid tumors) (une réponse partielle observée sur 341 patients). Comment expliquer ce succès alors que de très nombreux essais d’immunisation avec des cellules mononucléées ou des cellules dendritiques sensibilisées avec des antigènes avaient été réalisés antérieurement sans succès ? Il ne semble pas que la méthode d’immunisation soit innovante mais l’une des caractéristiques de cet essai est de n’inclure que des patients sans métastases viscérales, alors que la présence de métastases est souvent associée à une résistance à l’immunothérapie. Si ce premier vaccin thérapeutique redonne espoir aux immunothérapeutes du cancer après les très nombreux échecs enregistrés au cours de ces dernières années, l’impact clinique de ce produit risque d’être faible en raison de son coût (93 000 dollars US pour les trois injections du vaccin) et de la complexité de la préparation thérapeutique.
Autres succès de l’immunothérapie reposant sur des vaccins antitumoraux
Le groupe de P. Hwu a récemment montré dans un essai clinique de phase III randomisé qu’un vaccin antitumoral associant le peptide gp100 - antigène de différenciation mélanocytaire - à de l’IL-2 à fortes doses était plus efficace que l’administration d’IL-2 seule à fortes doses chez des patients atteints de mélanomes métastatiques [8]. Ainsi le taux de réponse clinique chez les patients qui recevaient le vaccin et l’IL-2 (n = 91) était de 16 % alors qu’il n’était que de 6 % en l’absence du vaccin (p = 0,03). La survie globale des patients ayant reçu le vaccin et l’IL-2 était de 17,8 mois contre seulement 11 mois chez les patients ayant reçu l’IL-2 seule (p = 0,06). Les deux groupes de patients étaient stratifiés en fonction de facteurs pronostiques connus pour éviter un biais dans l’analyse.
Les cellules de lymphomes folliculaires (lymphocytes B) expriment à leur membrane une immunoglobuline (Ig) composée d’une partie variable (idiotype2) propre à chaque patient et d’une partie constante. Il a été récemment montré dans un essai de phase III que les patients atteints de lymphomes folliculaires mis en rémission clinique après chimiothérapie et traités par un vaccin composé de l’idiotype du lymphome couplé à une protéine porteuse (KLH, keyhole limpet hemocyanin) et associé à du GM-CSF bénéficiaient d’une rémission prolongée (44,2 mois) par rapport aux patients traités uniquement avec la KLH et le GM-CSF (temps sans récidive 30,6 mois) (p = 0,047) [9]. Le temps de production de ce vaccin personnalisé étant long (6 à 12 mois), de nombreux patients inclus dans ce protocole ont rechuté avant de pouvoir bénéficier du vaccin et ont été exclus de l’étude pour l’analyse des résultats.
Par ailleurs dans deux essais cliniques de phase III, un traitement immunomodulateur par l’anticorps anti-CTLA-4 (cytotoxic T-lymphocyte antigen) ou ipilimumab a démontré son efficacité chez des patients atteints de mélanomes [10]. Ces résultats sont détaillés dans un article de C. Robert et C. Mateus dans ce même numéro [57].
Leçons des nombreux échecs de l’immunothérapie et de ses premiers succès
À l’exception du succès thérapeutique des anticorps en oncologie, les autres approches d’immunothérapie restent marginales dans l’arsenal thérapeutique des cancérologues. Pour améliorer les indications et l’efficacité de ces traitements, différents paramètres cliniques et biologiques devraient être pris en compte.
Sélection des patients : paramètres prédictifs de la réponse à l’immunothérapie
Aussi bien lors des traitements d’immunothérapie par l’IL-2 que par l’anticorps anti-CTLA-4, des réponses cliniques complètes et durables sont observées chez un sous-groupe de patients (5 % à 10 %) [10, 11]. Il est important de considérer la durabilité des réponses car elle n’est pas observée avec les autres thérapeutiques ciblées3. L’identification de ce groupe de patients permettrait d’augmenter l’impact clinique de ces traitements.
On peut mentionner quelques paramètres importants qui ont été corrélés à une résistance à l’immunothérapie.
Présence de métastases viscérales
Sur le plan clinique, la présence de métastases viscérales est un facteur de résistance aux cytokines recombinantes (IL-2 et IL-12) ainsi qu’aux vaccins antitumoraux [12–14].
Biomarqueurs de l’inflammation
Des dosages sériques de différents biomarqueurs liés à l’inflammation ont permis de prédire une réponse à l’immunothérapie. Ainsi, les patients qui présentent un syndrome inflammatoire reflété par des concentrations élevées d’IL-6 ou de CRP (C reactive protein, synthétisée par le foie et régulée par l’IL-6) et ceux chez qui on détecte un nombre élevé de lymphocytes TH17 circulants4 sont le plus souvent résistants à des traitements par cytokines recombinantes ou par vaccination antitumorale [15, 16]. Des résultats récents montrent que ce rôle prédictif de la CRP sur la réponse à l’immunothérapie l’est aussi pour la réponse à certains immunomodulateurs comme l’anticorps anti-CTLA-4 [17].
Biomarqueurs liés à l’angiogenèse
D’autres travaux suggèrent qu’au cours du développement des tumeurs, les lymphocytes ont des difficultés à s’infiltrer au cœur des cellules tumorales. Une néoangiogenèse constituée de vaisseaux anarchiques et inefficace sur le plan fonctionnel pourrait expliquer ce phénomène, et des antiangiogéniques améliorent l’infiltration des tumeurs par les cellules immunes [18]. Dans ce contexte, il est intéressant de noter que des concentrations élevées de VEGF, reflétant une néoangiogenèse importante, ont été associées à une résistance à l’immunothérapie [19].
Microenvironnement tumoral
Plus récemment, certains auteurs ont analysé le microenvironnement des tumeurs pour tenter de définir le profil de chimiokines nécessaires au recrutement des lymphocytes T CD8+. Dans ces études, la présence de certaines chimiokines (CCL2, CCL3, CCL4, CCL5, CXCL9, CXCL10, CX3CL1) était corrélée avec la densité d’infiltration des lymphocytes T CD8+ intratumoraux [20]. Le groupe de Thomas F. Gajewsky a montré que l’absence d’expression de ces chimiokines avant vaccination antitumorale corrélait avec un échec du traitement, suggérant peut-être un problème de homing des lymphocytes T induits [21].
Dissémination d’épitopes (epitope spreading)
Différents groupes ont montré chez des patients vaccinés avec un antigène tumoral donné l’induction d’une réponse lymphocytaire dirigée contre d’autres antigènes tumoraux [22]. Les mécanismes mis en jeu dans ce phénomène de dissémination épitopique ne sont pas bien élucidés : ils pourraient être secondaires à la lyse tumorale par les premiers lymphocytes T induits, favorisant la présentation croisée d’antigènes tumoraux libérés, ou à l’amplification (via la production locale de cytokines au décours de la vaccination) de clones T antitumoraux déjà présents dans le microenvironnement tumoral. La mise en évidence de cette dissémination épitopique est en général associée à une bonne réponse clinique [23].
Indications privilégiées de l’immunothérapie : lésions prénéoplasiques ou cancers à des stades précoces
À l’exception de l’essai de Hwu avec le vaccin gp100 associé à l’IL-2, les résultats les plus significatifs ont été obtenus en traitant des patients à des stades précoces de leur maladie ou en situation adjuvante - c’est-à-dire sans qu’il y ait de lésions tumorales visibles, dans le but d’éviter les récidives - ou ceux avec une maladie résiduelle. Ainsi, deux vaccins thérapeutiques contre le papillomavirus (HPV) ont été développés par la société Transgène et par le groupe de Cornelis J.M. Melief. Le vaccin de la société Transgène repose sur un virus recombinant dérivé de la vaccine atténuée (MVA) et contenant les ADNc codant pour les antigènes E6 et E7 de l’HPV16 et pour l’IL-2. Ce vaccin s’est révélé efficace dans le traitement de lésions dysplasiques sévères du col de l’utérus, les lésions disparaissant chez 30 % des patientes (7/21) 12 mois après l’administration du vaccin [24]. Une régression des lésions a aussi été observée par le groupe de Cornelis J.M. Melief chez 47 % des patientes atteintes de dysplasies vulvaires, un an après l’administration d’un vaccin composé de mélanges de longs peptides dérivés des protéines E6 et E7 de l’HPV [25]. De façon intéressante, ces mêmes vaccins n’ont pas démontré une efficacité clinique chez des patientes atteintes de cancer du col de l’utérus primaire ou métastatique [26].
Comme nous l’avons mentionné ci-dessus, un vaccin ciblant l’idiotype exprimé par les Ig des cellules B du lymphome folliculaire permettait de prolonger la durée de la rémission obtenue par chimiothérapie chez ces patients [9]. En revanche, un tel vaccin n’a pas d’efficacité si la sélection des patients inclut ceux qui sont en rémission partielle après chimiothérapie ou dont la maladie est stable ; cela suggère que ce vaccin est surtout efficace pour traiter la maladie résiduelle [27].
Par ailleurs, les résultats positifs de deux essais cliniques de phase II de vaccination antitumorale en situation adjuvante prouvent une meilleure efficacité des vaccins antitumoraux administrés à des stades précoces de la maladie. L’un de ces essais cliniques a été développé par la société GSK et repose sur l’administration de la protéine Mage A3 mélangée à un adjuvant chez des patients atteints de cancer du poumon [28]. L’autre essai est celui réalisé par le groupe de G.E. Peoples qui utilise un peptide dérivé d’Her2/neu comme immunogène pour vacciner des patientes atteintes de cancers du sein à haut risque de récidive [29]. Deux essais cliniques de phase III ont ensuite été lancés et sont en cours d’évaluation.
Trouver de nouveaux paramètres cliniques d’évaluation de la réponse à l’immunothérapie
Les critères standard d’évaluation clinique de la réponse aux traitements comme la réduction du volume tumoral par les critères RECIST ou WHO (World health organization) ne semblent pas adaptés à l’immunothérapie [30]. En effet, après immunothérapie, les taux de réponses objectives obtenus par ces critères standard sont assez modestes et ces paramètres d’évaluation sont progressivement abandonnés au profit de nouveaux critères comme la survie globale ou la survie sans progression (voir note 1). Contrairement à la réponse aux traitements conventionnels (chimiothérapie, radiothérapie, etc.), la réponse à l’immunothérapie peut être différée de plusieurs mois [31]. Dans l’essai de phase III avec le sipuleucel [11], les courbes de survie des patients du groupe contrôle et du groupe traité ne se dissocient qu’après plusieurs mois de traitement, ce qui réduit la puissance statistique des résultats.
Une augmentation transitoire des lésions (peut-être liée à l’infiltration des cellules immunes) peut même précéder leur régression [32]. Enfin, des réponses dissociées sont assez fréquentes : certaines lésions disparaissent de façon concomitante à l’apparition de nouvelles. Or, les critères RECIST considèrent l’apparition de nouvelles lésions comme un signe de progression de la maladie, même si, par ailleurs, une majorité des lésions ont régressé. Pour mieux prendre en compte ce type de réponse, on a proposé d’utiliser de nouveaux critères - IrRC : immune-related response criteria - comme la mesure du volume tumoral - incluant aussi bien les nouvelles que les anciennes lésions - considérée comme une variable continue [30].
Auto-immunité et réponse à l’immunothérapie
Les antigènes tumoraux étant souvent également exprimés par les cellules normales, il n’était pas surprenant qu’une réponse antitumorale puisse s’accompagner d’une réponse auto-immune. Les protocoles cliniques reposant sur le transfert de lymphocytes T antitumoraux, de cytokines recombinantes (IL-2, IFNα) ou d’anticorps immunomodulateurs comme l’anticorps anti-CTLA-4 (ipilimumab) s’accompagnent de manifestations biologiques et/ou cliniques auto-immunes le plus souvent bénignes (vitiligo, thyroïdite) mais parfois sévères (entérocolite auto-immune) notamment après administration de l’ipilimumab [58]. Dans un premier temps, des travaux ont rapporté une corrélation entre l’apparition de ces manifestations auto-immunes et la réponse clinique aux cytokines (IL-2 et IFNα) [12, 33]. Cette corrélation est aujourd’hui discutée car elle pourrait dépendre plus de la durée du traitement que de la réponse clinique [34]. En revanche, les manifestations auto-immunes observées après administration de l’anticorps anti-CTLA-4 sont plus précoces et restent corrélées à la réponse au traitement [35, 57] (→).
(→) Voir l'article de Caroline Robert et Christine Mateus, page 850 de ce numéro
L’immunothérapie : quelles perspectives ?
Améliorer l’immunogénicité des vaccins et mieux prendre en compte l’immunosuppression associée aux tumeurs
Améliorer les vaccins
En raison du rôle complexe (protumoral ou antitumoral) des lymphocytes B dans le contrôle du développement des tumeurs dans les modèles précliniques [36] et des échecs des vaccins thérapeutiques induisant une immunité humorale isolée, l’objectif des vaccins thérapeutiques est d’induire des lymphocytes T cytotoxiques dont l’activité antitumorale est bien démontrée. Néanmoins, le succès des anticorps thérapeutiques incite à reconsidérer ce dogme.
L’utilisation de peptides longs comprenant des épitopes peptidiques reconnus par les lymphocytes CD4+ et CD8+, de virus recombinants avec les ADNc codant pour des antigènes tumoraux ou de cellules dendritiques sensibilisées avec des lysats tumoraux ou des peptides, s’est avérée efficace pour induire des réponses lymphocytaires T CD8+ antitumorales dans des essais cliniques chez l’homme, qui sont parfois associées à des réponses cliniques. Néanmoins, chez des patients atteints de cancers métastatiques, cette induction lymphocytaire est souvent transitoire [37, 38]. Pour minimiser les risques d’immunisation contre le vecteur viral, des stratégies de type prime-boost utilisant deux vecteurs viraux recombinants différents codant pour le même antigène ont été développées avec un certain succès [31].
D’autres vecteurs non réplicatifs ciblant différentes sous-populations de cellules dendritiques sont actuellement développés par notre groupe et d’autres équipes [39–42]. Ces nouveaux outils associés à l’identification de nouveaux adjuvants pour augmenter l’immunité cellulaire devraient permettre de mieux amplifier la réponse immune antitumorale sur les plans qualitatif et quantitatif.
Inhiber l’immunosuppression associée aux tumeurs
Les premiers échecs des protocoles d’immunothérapie et une meilleure connaissance du microenvironnement tumoral ont suggéré l’hypothèse selon laquelle l’immunosuppression associée aux tumeurs pouvait expliquer la résistance des tumeurs à l’immunothérapie.
Ainsi, la tumeur est infiltrée par des lymphocytes T régulateurs (CD4+CD25+Foxp3+) ou des cellules myéloïdes suppressives (MDSC) (population hétérogène composée de progéniteurs myéloïdes et de cellules myéloïdes immatures) capables d’inhiber les fonctions effectrices des lymphocytes T et des cellules NK.
Les proportions respectives de lymphocytes T régulateurs et de cellules effectrices antitumorales lors de l’apparition de la tumeur détermineraient le rejet de la tumeur ou sa progression [43]. De même, le succès ou l’échec de protocoles de vaccination antitumorale pourrait dépendre du ratio entre les lymphocytes T effecteurs et régulateurs induits après vaccination [44].
Les mécanismes d’action de ces populations suppressives restent mal élucidés, et différentes molécules ont été évaluées pour leur capacité de les inhiber ou de les éliminer, mais sans qu’aucune stratégie ne soit encore validée sur le plan clinique. Par ailleurs, l’utilisation d’un anticorps anti-CTLA-4 se fonde justement sur la présence dans les tumeurs de lymphocytes T que l’expression de CTLA-4 - molécule de costimulation inhibitrice - a rendus anergiques [60]. D’autres molécules de costimulation inhibitrices (PD1, Tim-3, BTLA, etc., voir Glossaire) pourraient aussi contribuer à cette anergie des lymphocytes T, de même que la présence dans le microenvironnement tumoral de molécules suppressives (TGFβ, IL-10, IDO, voir Glossaire) (Figure 2) [45, 46]. Le blocage de ces molécules par des anticorps ou des petites molécules chimiques est en cours d’évaluation (Figure 2).
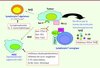 |
Figure 2. Principales stratégies développées pour inhiber l’immunosuppression associée au cancer. L’immunosuppression associée au cancer peut être secondaire au recrutement de cellules suppressives (lymphocytes T régulateurs, cellules myéloïdes suppressives) dans le microenvironnement des tumeurs sous l’influence de molécules sécrétées par la tumeur (TGFβ, VEGF, etc.). Par ailleurs, les lymphocytes T qui infiltrent les tumeurs sont peu fonctionnels et anergiques. Différents mécanismes peuvent expliquer cet état d’anergie : (1) expression par les lymphocytes T de molécules de costimulation inhibitrices (CTLA-4, PD1, Tim-3, etc.) qui, en interagissant avec leurs ligands exprimés sur les cellules tumorales ou les cellules stromales, vont déclencher un signal d’inhibition sur ces lymphocytes ; (2) des molécules produites par la tumeur (TGFβ, IL-10, IDO, etc.) peuvent inhiber la fonctionnalité des lymphocytes T. Différentes stratégies sont actuellement développées pour contrecarrer ces mécanismes d’inhibition (regroupées sur la figure dans les différents rectangles rouges). |
Certaines cytokines (IL-7, IL-15, IL-21, etc.) ou certains adjuvants comme des ligands de TLR (CpG) pourraient également lever l’état d’anergie des lymphocytes T infiltrant les tumeurs ou les rendre réfractaires aux mécanismes d’immunosuppression [46]. Toutefois, l’action ambivalente de certaines cytokines ou agonistes des TLR rend difficile leur manipulation sur le plan clinique [47, 48].
La variété des mécanismes d’immunosuppression est telle que l’échec d’approches d’immunothérapie pourrait paraître inévitable, surtout lors de cancers évolués où ces mécanismes sont dominants. Mais l’hypothèse est qu’un équilibre existe dans le stroma entre les mécanismes pro et antitumoraux, et que l’objectif de l’immunothérapie n’est pas d’éliminer tous les mécanismes suppresseurs mais de favoriser préférentiellement les réponses antitumorales.
Associer l’immunothérapie aux approches antitumorales conventionnelles
Chimiothérapie et immunothérapie
Jusqu’à ces dernières années, les chimiothérapies étaient uniquement vues comme un traitement antagoniste de l’immunothérapie en raison de leur rôle aplasiant, et notamment lymphopéniant. Les travaux pionniers des groupes de Laurence Zitvogel et Guido Kroemer ont remis en cause cette vision en montrant que certaines chimiothérapies (anthracyclines, oxiplatine, etc.) pouvaient entraîner une mort immunogénique qui active le système immunitaire et participe à l’efficacité clinique de la chimiothérapie. Une analyse moléculaire des conséquences de cette mort immunogénique aboutissant au rejet des tumeurs a montré que l’expression à la surface de la tumeur de la calréticuline, associée à la production par la cellule tumorale d’ATP et d’HMGB1, était responsable du recrutement de cellules de l’immunité innée, de l’activation de « l’inflammasome » et de la production de cytokines pro-inflammatoires et d’un signal de phagocytose des cellules tumorales par les cellules dendritiques [49, 58]. Par ailleurs, certaines chimiothérapies peuvent inhiber l’immunosuppression via leur action sur les lymphocytes T régulateurs ou les cellules myéloïdes suppressives et l’induction d’une augmentation de l’expression de ligands de NKG2D favorisant l’activation des cellules NK [50]. Chez l’homme, l’association d’une chimiothérapie et d’un vaccin viral recombinant avec l’ADNc codant pour Muc1 a donné des résultats encourageants [37].
Ces résultats importants ouvrent la voie à des associations thérapeutiques pertinentes entre certaines chimiothérapies et l’immunothérapie.
Radiothérapie et immunothérapie
Les effets cliniques d’une radiothérapie peuvent aussi dépendre de sa capacité à stimuler le système immunitaire, et son action synergique avec des vaccins antitumoraux ou des immunomodulateurs a été décrite [51]. Différents mécanismes peuvent l’expliquer : l’augmentation de l’expression des molécules de CMH de classe I par la tumeur, la facilitation de la présentation croisée d’antigènes tumoraux par les cellules dendritiques, le recrutement de lymphocytes T intratumoraux, la mort immunogénique de la cellule tumorale.
L’ensemble de ces données constitue un nouveau paradigme concernant les effets antitumoraux de la radiothérapie : on pourrait envisager d’une part de majorer son efficacité en l’associant à des thérapies immunostimulantes, et d’autre part d’appliquer une irradiation localisée pour majorer l’immunité antitumorale. La cinétique d’administration du vaccin par rapport à la radiothérapie, le fractionnement (dose par séance) et les doses totales de radiothérapie sont autant de paramètres importants pour obtenir cette synergie.
Antiangiogéniques et immunothérapie
Différents travaux suggèrent que l’inhibition de l’angiogenèse peut contrecarrer l’immunosuppression induite par les tumeurs. Ainsi, le VEGF sécrété par la tumeur bloque la maturation des cellules dendritiques et favorise l’expansion des lymphocytes T régulateurs [52]. L’hypoxie liée à la néoangiogenèse inefficace favorise aussi l’immunosuppression.
Chez l’homme, l’administration de sunitinib, un antiangiogénique qui inhibe la signalisation en aval du récepteur du VEGF, chez des patients atteints de cancer du rein a entraîné la diminution des lymphocytes T régulateurs et des MDSC [52] ainsi que le blocage d’autres mécanismes d’immunosuppression (Tableau II). Nous avons montré que la diminution des lymphocytes T régulateurs était associée à une meilleure survie des patients traités par le sunitinib [53, 54]. Cette capacité de certains traitements antiangiogéniques à lever l’immunosuppression associée au cancer, et les résultats synergiques encourageants observés dans les modèles précliniques avec l’immunothérapie [52, 55], constituent autant d’arguements en faveur de protocoles qui associent des molécules antiangiogéniques à des approches d’immunostimulation.
Exemples de mécanismes d’immunosuppression bloqués par certaines molécules antiangiogéniques.
Conclusion
Les récents succès cliniques d’approches d’immunothérapie ont redonné un dynamisme à cette voie thérapeutique. Une meilleure compréhension des mécanismes de résistance, une amélioration des vaccins et des immunomodulateurs et une meilleure sélection des patients pourront permettre d’augmenter encore l’impact clinique de ces traitements. La mise en évidence d’une synergie entre les traitements conventionnels utilisés en cancérologie (chimiothérapie, radiothérapie, traitement antiangiogénique, etc.) et l’immunothérapie est une opportunité pour mieux intégrer cette dernière dans les stratégies thérapeutiques actuelles en cancérologie.
Glossaire
ACE : antigène carcinoembryonnaire
ADCC : antibody dependent cell cytotoxicity
BTLA : B- and T-lymphocyte attenuator
CDK-4 : cyclin-dependent kinase 4
CTLA-4 : cytotoxic T-lymphocyte antigen
CRP : C reactive protein
EBV : Epstein-Barr virus
EGF : epithelial growth factor
GM-CSF : granulocyte-macrophage colony-stimulating factor
HBV : hepatitis B virus
HCV : hepatitis C virus
HMGB1 : high-mobility group protein B1
HPV : human papillomavirus
IDO : indoleamine 2,3-dioxygénase
IFN : interféron
KLH : keyhole limpet hemocyanin
LDH : lactate déshydrogénase
Mart 1 / Melan A : melanoma-associated antigen recognized by T cells
MDSC : myeloid derived suppressive cells
Muc1 : mucin 1
NK : natural killer
NKG2D : natural killer group 2D (ligands : MHC class I polypeptide-related sequence)
PAP : phosphatase acide prostatique
PD1 : programmed death 1 (membre de la famille CD28/CTLA-4)
PSA : prostate specific antigen
PSMA : prostate specific membrane antigen
RECIST : response evaluation criteria in solid tumors
TGFb : transforming growth factor β
Tim-3 : T cell immunoglobulin mucin-3
TLR : toll-like receptors
VEGF : vascular endothelial growth factor
Conflit d’intérêts
J. Y. Bonnefoy déclare avoir des liens durables et avoir participé à des interventions ponctuelles avec l’entreprise Transgene.
E. Tartour, F. Sandoval et W.H. Fridman déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Ce travail a été soutenu par l’ARC, la Ligue contre le cancer, le CIC-BT 505, le Cancéropôle, l’ANR, le fonds d’amorçage de l’AP-HP, le pôle de compétitivité Medicen, Labex Immuno-Oncology.
En cancérologie on distingue : (1) la survie globale : temps entre le début du traitement (cela peut être aussi le diagnostic de la maladie mais ici, c’est le début du traitement) et le décès et (2) la survie sans progression : temps entre le début du traitement et la date où la maladie recommence à évoluer, ce qui signifie un échec du traitement même si le patient est toujours vivant.
Déterminants idiotypiques : déterminants antigéniques présents sur les domaines variables des Ig, au niveau des CDR, régions hypervariables déterminant la complémentarité avec l’antigène, ou en dehors. Ces déterminants sont uniques pour chaque patient.
En terme de réponse clinique en cancérologie, on ne considère souvent que le critère lié à la diminution du volume tumoral : la réponse clinique est complète si on observe une disparition de toutes les lésions ciblées, partielle si la diminution est d’au moins 30 % du volume tumoral total (critères RECIST). La durabilité signifie la durée de la réponse. On peut avoir une réponse complète qui ne dure que 3 mois et une réponse complète qui dure des années. Avec l’immunothérapie, le taux de réponse sur le volume tumoral est souvent moins important qu’avec la chimiothérapie mais la durée des réponses (ou la persistance des réponses) est plus longue, ce qui est un élément important à prendre en compte pour le patient.
Lymphocytes T-CD4 produisant la cytokine IL-17 et favorisant l’inflammation.
Références
- Tartour E, Mathiot C, Fridman WH. Current status of interleukin-2 therapy in cancer. Biomed Pharmacother 1992 ; 46 : 473–484. [Google Scholar]
- Abes R, Gelize E, Fridman WH, et al. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood 2010 ; 116 : 926–934. [CrossRef] [PubMed] [Google Scholar]
- Taylor C, Hershman D, Shah N, et al. Augmented HER-2 specific immunity during treatment with trastuzumab and chemotherapy. Clin Cancer Res 2007 ; 13 : 5133–5143. [CrossRef] [PubMed] [Google Scholar]
- Bodmer A, Goetsch L, Favet L, et al. Anticorps et tumeurs solides : cibles établies et pistes prometteuses. Med Sci (Paris) 2009 ; 25 : 1090–1098. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Pages F, Galon J, Dieu-Nosjean MC, et al. Immune infiltration in human tumors : a prognostic factor that should not be ignored. Oncogene 2010 ; 29 : 1093–1102. [CrossRef] [PubMed] [Google Scholar]
- Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol 2009 ; 21 : 233–240. [CrossRef] [PubMed] [Google Scholar]
- Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010 ; 363 : 411–422. [CrossRef] [PubMed] [Google Scholar]
- Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med 2011 ; 364 : 2119–2127. [CrossRef] [PubMed] [Google Scholar]
- Schuster SJ, Neelapu SS, Gause BL, et al. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J Clin Oncol 2011 ; 29 : 2787–2794. [CrossRef] [PubMed] [Google Scholar]
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011 ; 364 : 2517–2526. [Google Scholar]
- Atkins MB. Treatment selection for patients with metastatic renal cell carcinoma : identification of features favoring upfront IL-2-based immunotherapy. Med Oncol 2009 ; 26 (suppl 1): 18–22. [CrossRef] [PubMed] [Google Scholar]
- Phan GQ, Attia P, Steinberg SM, et al. Factors associated with response to high-dose interleukin-2 in patients with metastatic melanoma. J Clin Oncol 2001 ; 19 : 3477–3482. [PubMed] [Google Scholar]
- Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004 ; 10 : 909–915. [Google Scholar]
- Haicheur N, Escudier B, Dorval T, et al. Cytokines and soluble cytokine receptor induction after IL-12 administration in cancer patients. Clin Exp Immunol 2000 ; 119 : 28–37. [CrossRef] [PubMed] [Google Scholar]
- Tartour E, Blay JY, Dorval T, et al. Predictors of clinical response to interleukin-2. Based immunotherapy in melanoma patients: a French multiinstitutional study. J Clin Oncol 1996 ; 14 : 1697–1703. [PubMed] [Google Scholar]
- Derhovanessian E, Adams V, Hahnel K, et al. Pretreatment frequency of circulating IL-17+ CD4+ T-cells, but not Tregs, correlates with clinical response to whole-cell vaccination in prostate cancer patients. Int J Cancer 2009 ; 125 : 1372–1379. [CrossRef] [PubMed] [Google Scholar]
- Marshall M, Ribas A, Huang B. Evaluation of baseline serum C-reactive protein (CRP) and benefit from tremelimumab compared to chemotherapy in first-line melanoma. ASCO meeting abstracts, June 14th, 2010. J Clin Oncol 2010 ; 28 : 2609 (abstract). [CrossRef] [Google Scholar]
- Shrimali RK, Yu Z, Theoret MR, et al. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res 2010 ; 70 : 6171–6180. [CrossRef] [PubMed] [Google Scholar]
- Sabatino M, Kim-Schulze S, Panelli MC, et al. Serum vascular endothelial growth factor and fibronectin predict clinical response to high-dose interleukin-2 therapy. J Clin Oncol 2009 ; 27 : 2645–2652. [CrossRef] [PubMed] [Google Scholar]
- Mlecnik B, Tosolini M, Charoentong P, et al. Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology 2010 ; 138 : 1429–1440. [CrossRef] [PubMed] [Google Scholar]
- Gajewski TF, Fuertes M, Spaapen R, et al. Molecular profiling to identify relevant immune resistance mechanisms in the tumor microenvironment. Curr Opin Immunol 2010 ; 23 : 286–292. [CrossRef] [PubMed] [Google Scholar]
- Germeau C, Ma W, Schiavetti F, et al. High frequency of antitumor T cells in the blood of melanoma patients before and after vaccination with tumor antigens. J Exp Med 2005 ; 201 : 241–248. [CrossRef] [PubMed] [Google Scholar]
- Butterfield LH, Ribas A, Dissette VB, et al. Determinant spreading associated with clinical response in dendritic cell-based immunotherapy for malignant melanoma. Clin Cancer Res 2003 ; 9 : 998–1008. [PubMed] [Google Scholar]
- Acres B, Bonnefoy JY. Clinical development of MVA-based therapeutic cancer vaccines. Exp Rev Vaccines 2008 ; 7 : 889–893. [CrossRef] [Google Scholar]
- Kenter GG, Welters MJ, Valentijn AR, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med 2009 ; 361 : 1838–1847. [CrossRef] [PubMed] [Google Scholar]
- Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer 2008 ; 8 : 351–360. [CrossRef] [PubMed] [Google Scholar]
- Freedman A, Neelapu SS, Nichols C, et al. Placebo-controlled phase III trial of patient-specific immunotherapy with mitumprotimut-T and granulocyte-macrophage colony-stimulating factor after rituximab in patients with follicular lymphoma. J Clin Oncol 2009 ; 27 : 3036–3043. [CrossRef] [PubMed] [Google Scholar]
- Brichard VG, Lejeune D. GSK’s antigen-specific cancer immunotherapy programme : pilot results leading to Phase III clinical development. Vaccine 2007 ; 25 (suppl 2): B61–B71. [CrossRef] [PubMed] [Google Scholar]
- Peoples GE, Gurney JM, Hueman MT, et al. Clinical trial results of a HER2/neu (E75) vaccine to prevent recurrence in high-risk breast cancer patients. J Clin Oncol 2005 ; 23 : 7536–7545. [CrossRef] [PubMed] [Google Scholar]
- Hoos A, Eggermont AM, Janetzki S, et al. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Inst 2010 ; 102 : 1388–1397. [CrossRef] [PubMed] [Google Scholar]
- Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol 2010 ; 28 : 1099–1105. [CrossRef] [PubMed] [Google Scholar]
- van Baren N, Bonnet MC, Dreno B, et al. Tumoral and immunologic response after vaccination of melanoma patients with an ALVAC virus encoding MAGE antigens recognized by T cells. J Clin Oncol 2005 ; 23 : 9008–9021. [CrossRef] [PubMed] [Google Scholar]
- Gogas H, Ioannovich J, Dafni U, et al. Prognostic significance of autoimmunity during treatment of melanoma with interferon. N Engl J Med 2006 ; 354 : 709–718. [CrossRef] [PubMed] [Google Scholar]
- Bouwhuis MG, Ten Hagen TL, Suciu S, et al. Autoimmunity and treatment outcome in melanoma. Curr Opin Oncol 2010 ; 23 : 170–176. [CrossRef] [Google Scholar]
- Attia P, Phan GQ, Maker AV, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol 2005 ; 23 : 6043–6053. [CrossRef] [PubMed] [Google Scholar]
- Nelson BH. CD20+ B cells : the other tumor-infiltrating lymphocytes. J Immunol 2010 ; 185 : 4977–4982. [CrossRef] [PubMed] [Google Scholar]
- Ramlau R, Quoix E, Rolski J, et al. A phase II study of Tg4010 (Mva-Muc1-Il2) in association with chemotherapy in patients with stage III/IV Non-small cell lung cancer. J Thorac Oncol 2008 ; 3 : 735–744. [CrossRef] [PubMed] [Google Scholar]
- Bercovici N, Haicheur N, Massicard S, et al. Analysis and characterization of antitumor T-cell response after administration of dendritic cells loaded with allogeneic tumor lysate to metastatic melanoma patients. J Immunother 2008 ; 31 : 101–112. [CrossRef] [PubMed] [Google Scholar]
- Leibundgut-Landmann S, Osorio F, Brown GD, et al. Stimulation of dendritic cells via the dectin-1/Syk pathway allows priming of cytotoxic T-cell responses. Blood 2008 ; 112 : 4971–4980. [CrossRef] [PubMed] [Google Scholar]
- Berraondo P, Nouze C, Preville X, et al. Eradication of large tumors in mice by a tritherapy targeting the innate, adaptive, and regulatory components of the immune system. Cancer Res 2007 ; 67 : 8847–8855. [CrossRef] [PubMed] [Google Scholar]
- Adotevi O, Vingert B, Freyburger L, et al. B Subunit of Shiga toxin-based vaccines synergize with (alpha)-galactosylceramide to break tolerance against self antigen and elicit antiviral immunity. J Immunol 2007 ; 179 : 3371–3379. [PubMed] [Google Scholar]
- Vingert B, Adotevi O, Patin D, et al. The Shiga toxin B-subunit targets antigen in vivo to dendritic cells and elicits anti-tumor immunity. Eur J Immunol 2006 ; 36 : 1124–1135. [CrossRef] [PubMed] [Google Scholar]
- Darrasse-Jeze G, Bergot AS, Durgeau A, et al. Tumor emergence is sensed by self-specific CD44hi memory Tregs that create a dominant tolerogenic environment for tumors in mice. J Clin Invest 2009 ; 119 : 2648–2662. [PubMed] [Google Scholar]
- Welters MJ, Kenter GG, van Steenwijk PJ, et al. Success or failure of vaccination for HPV16-positive vulvar lesions correlates with kinetics and phenotype of induced T-cell responses. Proc Natl Acad Sci USA 2010 ; 107 : 11895–11899. [CrossRef] [Google Scholar]
- Fourcade J, Sun Z, Benallaoua M, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med 2010 ; 207 : 2175–2186. [CrossRef] [PubMed] [Google Scholar]
- Derre L, Rivals JP, Jandus C, et al. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest 2010 ; 120 : 157–167. [CrossRef] [PubMed] [Google Scholar]
- Badoual C, Bouchaud G, Agueznay Nel H, et al. The soluble alpha chain of interleukin-15 receptor : a proinflammatory molecule associated with tumor progression in head and neck cancer. Cancer Res 2008 ; 68 : 3907–3914. [CrossRef] [PubMed] [Google Scholar]
- Cherfils-Vicini J, Platonova S, Gillard M, et al. Triggering of TLR7 and TLR8 expressed by human lung cancer cells induces cell survival and chemoresistance. J Clin Invest 2010 ; 120 : 1285–1297. [CrossRef] [PubMed] [Google Scholar]
- Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell 2010 ; 140 : 798–804. [CrossRef] [PubMed] [Google Scholar]
- Zitvogel L, Apetoh L, Ghiringhelli F, et al. Immunological aspects of cancer chemotherapy. Nat Rev Immunol 2008 ; 8 : 59–73. [CrossRef] [PubMed] [Google Scholar]
- Lee Y, Auh SL, Wang Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood 2009 ; 114 : 589–595. [CrossRef] [PubMed] [Google Scholar]
- Gabrilovich DI, Ishida T, Nadaf S, et al. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin Cancer Res 1999 ; 5 : 2963–2970. [PubMed] [Google Scholar]
- Adotevi O, Pere H, Ravel P, et al. A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. J Immunother 2010 ; 33 : 991–998. [CrossRef] [PubMed] [Google Scholar]
- Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res 2009 ; 15 : 2148–2157. [CrossRef] [PubMed] [Google Scholar]
- Huang KW, Wu HL, Lin HL, et al. Combining antiangiogenic therapy with immunotherapy exerts better therapeutical effects on large tumors in a woodchuck hepatoma model. Proc Natl Acad Sci USA 2010 ; 107 : 14769–14774. [Google Scholar]
- Abès R, Dutertre CA, Teillaud JL. Les anticorps : mieux les connaître pour mieux s’en servir. Med Sci (Paris) 2009 ; 25 : 1011–1019. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Robert C, Mateus C. Anticorps anti–CTLA 4 : une avancée thérapeutique majeure dans le traitement du mélanome métastatique. Med Sci (Paris) 2011 ; 27 : 850–858. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Apetoh L, Ghiringhelli F, Zitvogel L. La calréticuline détermine l’immunogénicité de la chimiothérapie et de la radiothérapie antitumorales. Med Sci (Paris) 2007 ; 23 : 257–258. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Rousseaux S, Reynoird N, Gaucher J, Khochbin S. L’intrusion des régulateurs de l’épigénome mâle dans les cellules somatiques cancéreuses. Med Sci (Paris) 2008 ; 24 : 735–741. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Olive D, le Thi S, Xerri L, et al. Rôle de CTLA-4 dans la cosignalisation négative du système immunitaire. Med Sci (Paris) 2011 ; 27 : 842–849. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des tableaux
Classification et quelques exemples d’antigènes tumoraux chez l’homme. Les antigènes (Ag) de différenciation sont spécifiques d’un tissu donné. Ils seront exprimés par les cellules normales et les cellules tumorales dérivées de ces tissus. Les antigènes du groupe « cancer testis » sont exprimés par les cellules tumorales et les cellules germinales [59]. L’absence d’expression de molécules HLA par les cellules germinales limite le risque d’auto-immunité en cas de réponse antitumorale ciblant ces antigènes. Le fait que 10 % des cancers soient associés à des virus peut permettre d’envisager une vaccination prophylactique contre certains cancers (cancers du foie, du col de l’utérus, etc.). Abréviations : voir Glossaire.
Exemples de mécanismes d’immunosuppression bloqués par certaines molécules antiangiogéniques.
Liste des figures
|
Figure 1. Mode d’action des anticorps. Certains anticorps (Ac) peuvent bloquer la liaison de ligands sur des récepteurs de facteurs de croissance ou provoquer un signal direct d’apoptose vers la cellule tumorale (exemple : anticorps anti-EGFR). La majorité des anticorps vont recruter des cellules effectrices (NK, macrophages) qui vont entraîner la lyse de la cellule tumorale par un mécanisme d’ADCC (antibody dependent cell cytotoxicity). La partie Fab (variable) de l’anticorps se fixe sur la cellule tumorale et la partie Fc de l’anticorps se fixe sur des récepteurs Fcγ activateurs (RFcγI, IIA, IIIA) à la surface des cellules effectrices. Ce pontage entre la cellule effectrice et la cellule tumorale par l’anticorps déclenche la cytotoxicité médiée par les cellules exprimant les RFc activateurs. Des cellules dendritiques peuvent également internaliser la cellule tumorale et présenter des peptides dérivés d’antigènes tumoraux aux lymphocytes T. |
| Dans le texte | |
|
Figure 2. Principales stratégies développées pour inhiber l’immunosuppression associée au cancer. L’immunosuppression associée au cancer peut être secondaire au recrutement de cellules suppressives (lymphocytes T régulateurs, cellules myéloïdes suppressives) dans le microenvironnement des tumeurs sous l’influence de molécules sécrétées par la tumeur (TGFβ, VEGF, etc.). Par ailleurs, les lymphocytes T qui infiltrent les tumeurs sont peu fonctionnels et anergiques. Différents mécanismes peuvent expliquer cet état d’anergie : (1) expression par les lymphocytes T de molécules de costimulation inhibitrices (CTLA-4, PD1, Tim-3, etc.) qui, en interagissant avec leurs ligands exprimés sur les cellules tumorales ou les cellules stromales, vont déclencher un signal d’inhibition sur ces lymphocytes ; (2) des molécules produites par la tumeur (TGFβ, IL-10, IDO, etc.) peuvent inhiber la fonctionnalité des lymphocytes T. Différentes stratégies sont actuellement développées pour contrecarrer ces mécanismes d’inhibition (regroupées sur la figure dans les différents rectangles rouges). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.