")
")
| Issue |
Med Sci (Paris)
Volume 22, Number 12, Décembre 2006
|
|
|---|---|---|
| Page(s) | 1015 - 1016 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/200622121015 | |
| Published online | 15 December 2006 | |
Effets pervers des cytokines sur le développement cérébral périnatal ?
Noxious effects of pro-inflammatory cytokines on perinatal brain development ?
1
Service de Neurologie pédiatrique, Laboratoire de neuropédiatrie, Département de pédiatrie, Université de Sherbrooke, CHU de Sherbrooke, 3001, 12 e Avenue Nord, Sherbrooke (Québec) J1J 1H1 Canada
2
Unité de Neuropathologie, Département d’anatomo-pathologie, CHU Brugmann-HUDERF, Université Libre de Bruxelles, Bruxelles, Belgique
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Environ 1 % des nouveau-nés, surtout prématurés, développent autour de la naissance des dommages cérébraux qui affectent de façon sévère et durable leurs capacités motrices, cognitives et comportementales. Ces dommages affectent la substance blanche (leucomalacies périventriculaires, LPV) et les neurones (nécrose neuronale sélective) des hémisphères cérébraux. Paradoxalement, en dépit de l’amélioration considérable des soins périnatals, l’incidence de la paralysie cérébrale s’accroît dans les pays industrialisés. Cette constatation remet en question certaines hypothèses physiopathologiques, notamment celle concernant l’importance attribuée à l’hypoxie-ischémie (H/I). Le rôle physiopathologique de l’H/I reste néanmoins indéniable, par exemple, dans le contexte, très fréquent, des infarctus cérébraux périnatals [1]. Plusieurs études épidémiologiques montrent l’association entre les chorioamniotites bactériennes et la survenue de lésions du système nerveux central (SNC). Par quel mécanisme ces infections nuisent-elles au développement cérébral ? Les infections directes du SNC ne semblent guère en cause. En revanche, se discute l’hypothèse d’un effet neurotoxique de médiateurs de l’inflammation, comme l’IL-1β et le TNF-α (tumor necrosis factor-α) (Figure 1).
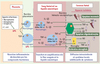 |
Figure 1. Hypothèse montrant le rôle des cytokines pro-inflammatoires dans les dommages cérébraux périnatals et la paralysie cérébrale subséquente. Les médiateurs de l’inflammation produits au niveau du site infecté seraient véhiculés par le sang et le liquide amniotique vers le système nerveux central dont la barrière hémato-encéphalique immature est permissive à certains composants de la réponse inflammatoire. |
Afin de vérifier ces hypothèses, nous avons d’abord étudié, directement dans le SNC humain, la possible association temporospatiale entre les lésions, et les cytokines pro-inflammatoires, leurs voies de signalisation, et l’activation en aval d’autres médiateurs neurotoxiques. En utilisant une banque de cerveaux humains, issus de nouveau-nés décédés après quelques jours de vie, et présentant des LPV, l’expression d’IL-1β et de TNF-α a été détectée à la surface des astrocytes, microglies, oligodendrocytes immatures sélectivement au sein de la substance blanche lésée et des neurones adjacents [2, 3]. L’expression d’IL-1β et de TNF-α est maximale à la phase précoce des LPV. L’étude de l’expression des récepteurs du TNF-α, p55TNF-α R (récepteur p55 du TNF-α) et p75 TNF-α R (récepteur p75 du TNF-α), appuie l’hypothèse d’un effet biologique du TNF-α, en montrant l’expression sélective de ces récepteurs au sein des LPV [4]. Le récepteur p75 TNF-α R est détecté principalement à la surface des cellules endothéliales, et, de façon moindre, sur les cellules gliales, tandis que l’expression de p55 TNF-α R est essentiellement gliale. L’interaction TNF-α/TNF-αR étant connue pour induire, d’une part, l’activation de la synthèse de NO (monoxyde d’azote) et, d’autre part, l’apoptose, il était intéressant d’étudier ces deux phénomènes biologiques. La démonstration de l’activation de la NO synthase et de l’apoptose, colocalisées au sein des LPV avec l’expression des TNF-α R, fournit un argument supplémentaire en faveur du rôle pathogène des cytokines [4]. Le récepteur p55 TNF-α R possède dans sa partie intracellulaire un domaine spécialisé dit de « mort cellulaire » dont l’activation déclenche une cascade apoptotique. Le rôle pathogène de p75 TNF-α R est illustré expérimentalement par l’observation d’une inflammation et d’une ischémie microvasculaire, indépendante de p55TNF-α R, dans le cerveau de souris transgéniques hyperexprimant TNF-α et p75TNF-α R humains [5]. Par ailleurs, p75 TNF-α R semble coopérer avec p55 TNF-α R en formant des hétérocomplexes ou en potentialisant l’effet pro-apoptotique de p55TNF-αR.
Plusieurs modèles de lésions cérébrales infligées à des animaux adultes illustrent déjà la neurotoxicité des cytokines, mais il n’est pas prouvé que ces phénomènes puissent s’extrapoler au SNC en développement. Les cytokines sont d’ailleurs également connues pour les rôles trophiques qu’elles exercent au cours du développement du SNC [6]. Afin d’éclaircir cette situation, nous avons mis au point un modèle de lésions cérébrales induites chez le rat par la combinaison d’une infection anténatale (injection intrapéritonéale de lipopolysaccharide [LPS] à la femelle gestante) et d’une H/I post-natale immédiate, agressions composites analogues à celles affectant le SNC humain [7]. Des lésions affectant la substance grise et la substance blanche, particulièrement dans les régions frontales contrôlant la motricité, sont induites par ces conditions expérimentales. L’effet délétère maximal est constaté avec la combinaison LPS+H/I par rapport au LPS ou à l’H/I seuls. L’étude de la réponse inflammatoire intracérébrale montre l’induction de la synthèse d’IL-1β, dont le pic de synthèse est observé après exposition combinée au LPS+H/I. La neurotoxicité de l’IL-1β a déjà été attestée dans plusieurs modèles animaux adultes de pathologies H/I mais reste à prouver à un stade précoce de développement [8]. L’IL-1β intervient dans un système interactif composé de plusieurs membres dont les principaux sont l’IL-1α qui, comme l’IL-1β, entre en compétition avec un antagoniste naturel, l’IL-1RA (interleukine-1 récepteur antagoniste), pour se lier à l’IL-1R1 (interleukine-1 récepteur de type 1), permettant la transduction du signal, et à l’IL-1 R2 (interleukine-1 récepteur de type 2, récepteur leurre sans effet biologique). Dans ce système complexe, il est évident que les données concernant un seul membre sont loin de refléter l’effet net global. Afin d’approcher la question de l’effet net de l’IL-1β, les cinétiques d’expression intracérébrale d’IL-1β, d’IL-1RA et de NF-kB (facteur nucléaire-κB) ont été étudiées en parallèle. De façon inhabituelle, par rapport aux données chez les adultes, un découplage entre l’expression d’IL-1β et d’IL-1RA est observé dans le SNC immature exposé au LPS et/ou à l’H/I, avec une chute de l’expression d’IL-1RA contrastant avec l’augmentation d’IL-1β, et ce dans toutes les conditions expérimentales mais avec un effet différentiel maximal lors de l’exposition au LPS+H/I. Cette dissociation est susceptible d’accroître les effets de l’IL-1β, ce qui est validé par la mise en évidence d’une augmentation de la synthèse intracérébrale de NF-κB dont l’ampleur est corrélée au niveau de découplage IL-1β/IL-1RA [9].
La neurotoxicité de l’IL-1β et du TNF-α doit encore être confirmée par l’étude de l’effet protecteur de l’inhibition de la synthèse ou d’une autre forme d’interférence dans l’action de ces cytokines, ce qui ouvrirait de nouvelles perspectives thérapeutiques anti-inflammatoires de neuroprotection pour les nouveau-nés.
Références
- Sébire G, Tabarki B, Saunders DE, et al. Cerebral venous sinus thrombosis in children : risk factors, presentation, diagnosis and outcome. Brain 2005; 128 : 477–89. [Google Scholar]
- Kadhim H, Tabarki B, Verellen G, et al. Inflammatory cytokines in the pathogenesis of periventricular leukomalacia. Neurology 2001; 56 : 1278–84. [Google Scholar]
- Kadhim H, Tabarki-Mlaiki B, De Prez C, et al. Cytokine immunoreactivity in cortical and subcortical neuronal centers in periventricular leukomalacia. Acta Neuropathol 2003; 105 : 209–16. [Google Scholar]
- Kadhim H, Khalifa M, Deltenre P, et al. Insight into the molecular network involved in cell death in periventricular leucomalacia. Neurology 2006; 67 : 293–9. [Google Scholar]
- Akassoglou K, Douni E, Bauer J, et al. Exclusive tumor necrosis factor (TNF) signaling by the p75TNF receptor triggers inflammatory ischemia in the CNS of transgenic mice. Proc Natl Acad Sci USA 2003; 100 : 709–14. [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Innate immunity : the missing link in neuroprotection and neurodegeneration. Nat Neurosci 2002; 3 : 216–27. [Google Scholar]
- Larouche A, Roy M, Kadhim H, et al. Neuronal injuries induced by perinatal hypoxic-ischemic insults are potentiated by prenatal exposure to LPS : animal model for perinatal acquired encephalopathy. Dev Neurosci 2005; 27 : 134–42. [Google Scholar]
- Pinteaux E, Rothwell N, Boutin H. Neuroprotective actions of endogenous interleukin-1 receptor antagonist (IL-1RA) are mediated by glia. Glia 2006; 53 : 551–6. [Google Scholar]
- Girard S, Larouche A, Gobeil F, Sarret P, Sébire G. Antenatal bacterial infection and/or immediately postnatal hypoxia-ischemia (HI) trigger early and prolonged intracerebral IL-1β synthesis. Ann Neurol 2006; 60 : 515–8. [Google Scholar]
© 2006 médecine/sciences - Inserm / SRMS
Liste des figures
|
Figure 1. Hypothèse montrant le rôle des cytokines pro-inflammatoires dans les dommages cérébraux périnatals et la paralysie cérébrale subséquente. Les médiateurs de l’inflammation produits au niveau du site infecté seraient véhiculés par le sang et le liquide amniotique vers le système nerveux central dont la barrière hémato-encéphalique immature est permissive à certains composants de la réponse inflammatoire. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.