")
")
| Issue |
Med Sci (Paris)
Volume 21, Décembre 2005
Métabolisme glucidolipidique et risque cardiovasculaire: Nouvelle approches
|
|
|---|---|---|
| Page(s) | 66 - 68 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20052111s66 | |
| Published online | 15 November 2005 | |
Les médicaments orphelins en Europe
Orphan medicinal products in Europe
Représentant français au comité des médicaments orphelins (EMEA), Service de Médecine Interne, CHNO des Quinze-Vingts, 28, rue de Charenton, 75012 Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Cet article explicite les mesures incitatives de la législation européenne de 1999 pour le développement des médicaments orphelins, et en dresse un bilan à 5 ans
Abstract
Incentives provided by the 1999 European regulation for the development of orphan drugs are described, and a picture of their outcome 5 years after is shown.
© 2005 médecine/sciences - Inserm / SRMS
Définition
Les médicaments dits orphelins sont en effet privés de sponsors car, s’adressant au traitement des maladies rares, la rentabilité incertaine de leur développement n’incite pas les laboratoires pharmaceutiques à les fabriquer. Dans la législation européenne, on définit les maladies rares par une prévalence inférieure à 5 pour 10 000 personnes au sein de la population européenne. On dénombre actuellement plus de 5 000 maladies rares, dont on estime qu’elles touchent au total entre 25 et 30 millions d’Européens pour une population actuelle de 454 millions (Eurostat 2004). La plupart de ces maladies sont graves, parce qu’elles menacent plus ou moins précocement le pronostic vital et/ou par le handicap qu’elles procurent. Au vu de ces chiffres, on comprend que le développement des médicaments orphelins est un véritable enjeu de santé publique.
Législation européenne
L’Union Européenne (UE) a tardé à apporter des mesures incitatives pour le développement des médicaments orphelins. Les États-Unis ont été les premiers à le faire avec l’Orphan Drug Act en 1983, suivis par le Japon en 1993, Singapour en 1997 et l’Australie en 1998. Largement inspiré par la législation américaine, le règlement 141/2000 sur les médicaments orphelins [1] a été voté par le parlement européen et le conseil des ministres le 16 décembre 1999, publié au Journal des Communautés Européennes le 22 janvier 2000, et il est entré en application le 27 avril 2000. Ce règlement destiné à stimuler l’émergence de médicaments pour le traitement des maladies rares en Europe, précise les critères de « désignation » d’un médicament orphelin et les mesures incitatives en direction de leurs sponsors.
La « désignation » d’un médicament orphelin correspond à la reconnaissance officielle de ce statut pour un produit, et permet aux sponsors de bénéficier des mesures incitatives accordées par le règlement pour les développer. L’évaluation des produits candidats est effectuée par un comité scientifique créé en mars 2000 au sein de l’EMEA, le Committee for Orphan Medicinal Products (COMP), qui se réunit mensuellement à cette intention. Celui-ci est composé d’un président et de 31 membres : un représentant de chaque État membre de l’UE, 3 membres de l’EMEA (European Agency for the Evaluation of Medicinal Products) et (innovation notable) trois représentants des associations de patients. Pour qu’un produit soit désigné « orphelin », le promoteur doit établir qu’il est dévolu au diagnostic, à la prévention, ou au traitement d’une maladie :
potentiellement létale ou génératrice d’un handicap chronique ;
affectant moins de 5 habitants de l’UE sur 10 000 ;
pour laquelle il n’y a pas de traitement ou, s’il en existe déjà, le sponsor doit apporter des arguments en faveur d’un bénéfice significatif attendu du nouveau produit.
Les produits sont soumis à des stades variables, mais souvent très précoces, de leur développement. Il peut arriver qu’il n’y ait encore aucune donnée clinique dans un dossier de « désignation », mais seulement des données sur modèle animal, voire in vitro. La « plausibilité médicale » du produit candidat est donc un élément constant des procédures d’évaluation. La philosophie du règlement étant de favoriser la mise à disposition de nouveaux médicaments pour les patients, il ne s’agit pas de bloquer l’émergence de nouveaux produits mais de s’assurer que ceux-ci aient le maximum de chances d’obtenir in fine leur autorisation de mise sur le marché (AMM). La « plausibilité » fait référence au rationnel de l’utilisation du produit dans l’indication considérée et, s’il s’adresse à un sous-type particulier d’une maladie, à la justification précise de cette restriction (par exemple : indication du Glivec® dans les tumeurs malignes stromales gastro-intestinales kit [CD 117] positives). Un autre sujet de discussion fréquent porte sur la définition de la maladie ciblée par le sponsor, qui doit correspondre à une entité médicale bien définie, cliniquement pertinente, et non à une subdivision arbitraire d’une maladie fréquente dont l’objectif premier est généralement de satisfaire le critère de prévalence.
La désignation des médicament orphelins est décidée par la Commission européenne sur les recommandations scientifiques du COMP et est suivie par une consignation dans le registre européen des médicaments orphelins.
Concernant les mesures incitatives pour les sponsors, elles comprennent une réduction de 50 % des taxes dues à l’EMEA, une aide méthodologique gratuite (protocol assistance) au sein de l’EMEA jusqu’à la constitution du dossier d’AMM, et (incitation in fine la plus importante) une exclusivité commerciale de 10 ans à compter de l’AMM, qui n’est donnée que pour les médicaments autorisés sur tout le territoire de l’UE (ce qui est automatique en cas de « procédure centralisée », déposée à l’EMEA. Cette procédure sera obligatoire à partir de novembre 2005).
2000-2005 : 5 ans de règlement européen sur les médicaments orphelins
La réglementation a largement joué son rôle de stimulation de la recherche et du développement de médicaments orphelins puisque d’avril 2000 à avril 2005, 458 dossiers ont été soumis et 260 produits ont été désignés « médicaments orphelins », soit un taux d’opinions positives de 56,7 % (à peu près similaire au taux de désignation par la FDA (Food and Drug Administration) aux États-Unis qui est légèrement supérieur à 60 %). Cette stimulation apparaît aussi dans le fait que 40 à 50 % des sponsors sont des petites et moyennes entreprises qui se sont investies dans ce « créneau », du fait d’incitations jugées suffisantes. La Figure 1 illustre le bilan de ces 5 années de législation européenne, depuis les candidatures à « désignation » orpheline jusqu’à l’AMM, déjà obtenue pour 22 produits détaillés dans le Tableau I. Ces nouvelles AMM concernent potentiellement plus d’un million de patients à travers l’Europe.
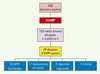 |
Figure 1. Médicaments orphelins en Europe : 2000-2005. |
Médicaments orphelins autorisés en Europe. *AMM par reconnaissance mutuelle (non européenne d’emblée). Seuls les 20 médicaments ayant une AMM dans toute l’Europe bénéficient de l’exclusivité commerciale de 10 ans.
Les produits « désignés » concernent plus de 200 maladies rares, majoritairement dans les domaines de la cancérologie (36 %), de l’immunologie (11 %), des maladies métaboliques (11 %), du système cardiovasculaire et respiratoire (10 %) et neuromusculaires ou osseuses (8 %). Dans 90 % des cas, ces produits concernent des maladies ayant une prévalence inférieure à 3/10 000 et dans 43 % des cas inférieure à 1/10 000. Une proportion significative comporte un usage pédiatrique : 11 % uniquement pédiatrique, et 43 % à usage adulte et pédiatrique. Plus de la moitié des produits désignés (53 %) sont véritablement innovants, dont 92 (21 %) sont des produits des biotechnologies, dont les thérapies émergeantes (anti-sens, géniques et cellulaires).
Les médicaments orphelins représentent désormais une proportion notable des AMM centralisées, délivrées à l’EMEA par le Committee for Medicinal Products for Human Use (CHMP), puisqu’en 2004 ils concernaient 6 des 34 (18 %) produits autorisés.
Mise à disposition des médicaments orphelins aux patients
Les États membres de l’UE ont différents systèmes de soins qui engendrent au mieux des dates variables d’apparition des nouveaux médicaments selon les pays, et au pire leur indisponibilité pure et simple dans certains d’entre eux. De nombreux paramètres interviennent : détermination du prix du médicament dans chaque pays sur lequel influe, entre autres, une taxation variable (jusqu’à 70 % d’écart de coût de traitement annuel par patient entre certains pays), des circuits de distribution, le niveau de priorité donné à la prise en charge des maladies rares, et enfin le problème crucial du remboursement de ces médicaments souvent très onéreux. En résumé après une AMM européenne, les médicaments orphelins n’apparaissent généralement pas dans tous les pays de l’UE, et le font avec des délais et à des prix très variables.
Une enquête a été menée en décembre 2004 par EURORDIS (European Organisation for Rare Disorders, coalition de plus de 200 associations de maladies rares) sur la disponibilité des 12 médicaments orphelins autorisés au niveau de l’UE avant le 31 décembre 2003, soit au moins 1 an plus tôt. Alors que le délai légal maximum de mise sur le marché après une AMM est de 180 jours, l’enquête montrait une médiane de disponibilité de 5 médicaments sur 12 dans les différents pays (allant de 100 % au Danemark, 92 % en Autriche et France, à 0 % en Lettonie et Lituanie).
Dans le souci d’améliorer la transparence par rapport à la mise à disposition aux patients des médicaments autorisés, le règlement européen 726/2004 du 31 mars 2004 exige des sponsors qu’ils rendent compte à l’EMEA, en sus des données de pharmacovigilance, du volume de leurs ventes par pays et de toute donnée relative aux prescriptions.
Perspectives
La dynamique « orphelins » semble bien enclenchée en Europe. Une enquête récente de l’EMEA auprès des sponsors indique qu’un tiers des médicaments « désignés » seraient au stade final de développement, et 40 % de ces sponsors pensent pouvoir soumettre un nouveau dossier de « désignation » dans les trois ans. Dans un tout récent rapport à la commission européenne [2], le COMP souligne néanmoins le besoin d’encourager davantage les États membres à adopter une politique nationale plus incitative pour le développement de médicaments orphelins, ainsi qu’à œuvrer vers plus de transparence et de coordination pour la mise à disposition des nouveaux produits aux patients.
Bien entendu, la prise en charge des maladies rares ne se limite pas à l’existence et à la dispensation d’un traitement. Améliorer la rapidité et l’exactitude des diagnostics, la prise en charge psychologique et sociale des patients et de leur famille, sont des éléments cruciaux mais hautement dépendants des politiques nationales pour leur mise en œuvre. Certains pays européens montrent la voie dans ce domaine, en particulier la France à travers le « Plan National Maladies Rares 2005-2008 » qui a notamment donné les moyens concrets de l’ouverture de « centres de référence » pour la prise en charge des maladies rares sur tout le territoire. Une amplification de ces politiques, européenne et nationales, est cependant indispensable pour que le vœu émis dans le préambule du règlement européen : « patients suffering from rare conditions should be entitled to the same quality of treatment than other patients », se rapproche de la réalité.
Références
- Regulation (EC) N0 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products, 2000, OJ, L018, 1–5. [Google Scholar]
- COMP Report to the Commission in Relation to article 10 of Regulation 141/2000 on Orphan Medicinal Products (July 2005). www.emea.eu.int [Google Scholar]
Liste des tableaux
Médicaments orphelins autorisés en Europe. *AMM par reconnaissance mutuelle (non européenne d’emblée). Seuls les 20 médicaments ayant une AMM dans toute l’Europe bénéficient de l’exclusivité commerciale de 10 ans.
Liste des figures
|
Figure 1. Médicaments orphelins en Europe : 2000-2005. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.