")
")
| Issue |
Med Sci (Paris)
Volume 21, Décembre 2005
Métabolisme glucidolipidique et risque cardiovasculaire: Nouvelle approches
|
|
|---|---|---|
| Page(s) | 12 - 15 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20052111s12 | |
| Published online | 15 November 2005 | |
Le lysosome a cinquante ans
The lysosome turns fifty
Prix Nobel de Médecine (1974), est professeur émérite à l’Université catholique de Louvain, Belgique, et à l’Université Rockefeller, New York, États-Unis
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Au cours d’une investigation visant à caractériserla glucose 6-phosphatase hépatique, le hasard d’une expérience contrôle révéla que la phosphatase acide, qui n’a aucun rapport avec la précédente, se trouvait sous forme latente et liée à des particules dans des homogénats fraîchement préparés de foie de rat. Des recherches entreprises pour élucider cette curieuse observation conduisirent, il y a 50 ans, à la découverte des lysosomes. Ces organites n’auraient pas pu être découverts si un strict respect du programmede recherche établi avait été exigé.
Abstract
In the course of an investigation aimed at characterizing hepatic glucose 6-phosphatase, the unrelated acid phosphatase of rat liver was serendipitously observed to be latent and particle-bound in freshly prepared homogenates. Experiments designed to elucidate this intriguing finding led, 50 years ago, to the discovery of lysosomes. This could not have happened if strict adherence to a previously set programme had been mandatory.
© 2005 médecine/sciences - Inserm / SRMS
Le mot « lysosome », du grec signifiant «corpuscule digestif», apparut pour la première fois dans une publication, il y a 50 ans [1]. Le choix du mot était loin d’être heureux, car le terme « lysozyme » – nom donné par Alexander Fleming, le célèbre découvreur de la pénicilline, à une enzyme bactériolytique qu’il avait identifiée – était en usage depuis 1921. Je croyais que les biologistes sauraient faire la différence entre les racines grecques sôma et zymê. Je me trompais car, aujourd’hui encore, il advient qu’on m’attribue à tort ce qui est dû à Fleming.
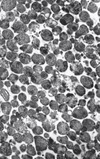 |
Figure 1. Lysosomes purifiés provenant de foie de rat (x10400). Reproduit du Journal of Cell Biology (1965; 26: 219-43), avec la permission de la Rockefeller University Press. |
À part mon manque de discernement comme inventeur de mots, j’étais à cette époque la personne la moins bien placée pour découvrir l’objet de mes efforts linguistiques. Comme je l’ai rappelé dans une rétrospective antérieure plus détaillée [2], mon ignorance de la cellule était abyssale et l’intérêt que je portais à son organisation était limité à ce qu’il fallait pour qu’un hépatocyte réponde à l’insuline. Mon domaine de compétence en tant que biochimiste était le métabolisme des hydrates de carbone et ma seule ambition était d’élucider le mécanisme d’action de l’insuline sur le foie [3]. De retour en Belgique en 1948, après des stages post-doctoraux en Suède et aux États-Unis, je formai un petit groupe composé d’un jeune médecin, Géry Hers, et de deux étudiants en médecine, Jacques Berthet et sa future femme, Lucie Dupret. Notre but était de clarifier l’action hépatique de l’insuline dont le caractère élusif prêtait à l’époque à de vives controverses.
Un heureux hasard ouvre une voie nouvelle
Notre intérêt se porta tout d’abord sur une enzyme hépatique, connue alors seulement en tant qu’hexose phosphatase, que je soupçonnais de pouvoir entraver l’action de l’insuline sur le tissu hépatique isolé. Nous pûmes identifier l’enzyme comme étant une glucose 6-phosphatase spécifique et trouvâmes, en outre, qu’elle était associée à une structure cellulaire précipitable en milieu acide [4].
Le hasard a voulu que cette découverte fut faite à un moment où la centrifugation fractionnée commençait tout juste à être utilisée pour étudier la localisation cellulaire des enzymes, principalement dans le tissu hépatique. Mise au point par Albert Claude [5, 6], au Rockefeller Institute for Medical Research (aujourd’hui Université Rockefeller) à New York, cette technique avait été perfectionnée par ses anciens collaborateurs George Palade et, surtout, George Hogeboom et Walter Schneider, qui furent pendant des années les vrais pionniers de ce domaine. La méthode mise au point par ces chercheurs commençait par un broyage modéré du tissu en présence de sucrose isotonique – suffisant pour démembrer les cellules tout en infligeant un minimum de dégats aux fragiles structures intracellulaires – menant à un mélange complexe de composants particulaires et solubles improprement appelé « homogénat ». Celui-ci était ensuite fractionné par des centrifugations successives à vitesse de rotation croissante. On séparait habituellement quatre fractions : une fraction nucléaire, une fraction de gros granules contenant surtout des mitochondries, une fraction de petits granules, appelés microsomes par Claude, et un surnageant final. Je connaissais ce procédé pour avoir rendu visite à Albert Claude avant mon retour des USA. J’avais gardé une forte impression de cette visite. Ceci a certainement compté dans notre décision d’adopter cette nouvelle technique – investissement important en temps et en travail pour notre petite équipe – dans le but d’identifier la structure à laquelle la glucose 6-phosphatase semblait être attachée. Je pensais que cette information serait utile pour nos travaux sur l’insuline.
Cette décision se révéla particulièrement fructueuse, mais pas dans le sens que j’attendais. Nous atteignîmes bien notre but, trouvant notamment que la glucose 6-phosphatase est associée aux microsomes [7]. Ce à quoi s’ajoutèrent des observations indiquant que l’enzyme devait être fermement liée à des membranes [8], anticipant ainsi la découverte, faite ultérieurement par d’autres, que les microsomes sont constitués principalement de fragments de réticulum endoplasmique. Mais le filon le plus précieux vint d’un résultat qui n’avait aucun rapport avec notre projet de recherche. Dans notre étude biochimique, une partie de notre travail avait été de différencier la glucose 6-phosphatase d’une autre phosphatase hépatique, la phosphatase acide non spécifique. Nous mesurâmes donc aussi cette enzyme dans nos fractions. À notre grande surprise, la phosphatase acide avait en grande partie disparu, ne montrant que des activités très faibles dans l’homogénat et dans les fractions obtenues. Lorsque les dosages furent refaits après 5 jours de conservation des préparations au réfrigérateur, les activités étaient remontées à des valeurs normales, l’augmentation la plus importante s’observant dans la fraction mitochondriale [9].
Cet étrange comportement d’une enzyme était trop curieux pour rester sans explication. La solution vint de recherches où des procédés divers étaient utilisés pour activer l’enzyme et qui indiquèrent – sur la base de données strictement biochimiques (à l’exclusion de toute étude morphologique) – que la phosphatase acide devait être enfermée dans des particules en forme de sac, entourées par une membrane qui empêchait la sortie de l’enzyme aussi bien que l’entrée du substrat. Le broyage vigoureux que nous pratiquions précédemment, de même que tout autre traitement physique susceptible de léser la membrane – y compris le simple vieillissement des préparations pendant quelques jours à basse température – permettait à l’enzyme d’interagir avec le substrat et supprimait ce que nous appelions sa « latence ».
Mais ceci n’était qu’une part du mystère. Non seulement l’enzyme était latente, mais sa répartition était curieuse: 2/3 de l’activité liée à des particules se retrouvant dans la fraction des mitochondries et 1/3 dans celle des microsomes. Au lieu de simplement entériner ce fait, nous tentâmes d’en comprendre la signification en appliquant divers schémas de centrifugation, pour arriver finalement à la conclusion que la phosphatase acide doit être associée à un groupe spécial de particules, différentes tant des mitochondries que des microsomes. Sur la base de ces résultats, nous établîmes un protocole de centrifugation permettant de séparer une petite fraction intermédiaire particulièrement riche en ces nouvelles particules – appelée L pour mitochondries légères (le mot lysosome n’existait pas encore) - encadrée par des fractions contenant le gros des mitochondries (M) et des microsomes (P). Dans ce travail, des enzymes furent utilisées pour la première fois comme marqueurs des particules intracellulaires auxquelles elles appartiennent: cytochrome-oxydase pour les mitochondries, glucose 6-phosphatase pour les microsomes. Ce procédé s’est avéré extrêmement précieux dans tous nos travaux ultérieurs.
Le découverte des lysosomes
Une fois ces résultats obtenus, nous étions accrochés. L’insuline et le métabolisme des hydrates de carbone furent abandonnés – sauf par Hers qui persista dans cette voie et apporta des contributions importantes à notre connaissance du métabolisme du fructose et du glycogène, jusqu’à ce qu’un autre heureux hasard le ramène aux lysosomes vingt ans plus tard (voir ci-dessous). Ma petite équipe, qui s’agrandissait lentement – avec l’arrivée de trois nouveaux étudiants en médecine, Françoise Appelmans, Henri Beaufay et Robert Wattiaux, et de deux postdoctorants, Robert Gianetto du Canada et Burton Pressman des États-Unis – entreprit de chercher systématiquement les enzymes qui, comme la phosphatase acide, étaient concentrées dans la fraction L. Quand vint 1955, nous avions déjà trouvé cinq nouvelles enzymes de ce type. Quatre d’entre elles étaient, comme la phosphatase acide, des hydrolases avec un pH optimum acide: une β-glucuronidase, une cathepsine (une protéase connue à présent sous le nom de cathepsine D), une ribonucléase et une désoxyribonucléase. La cinquième était une enzyme oxydante, l’urate-oxydase. Les quatre premières partageaient avec la phosphatase acide la propriété de latence [1, 12] et semblaient situées dans les mêmes particules. L’urate-oxydase n’était pas latente et se révéla finalement appartenir à un autre nouveau type d’organites cytoplasmiques, que nous avons appelés peroxysomes [13].
La présence, dans de petits sacs limités par une membrane, de cinq hydrolases acides agissant sur des substrats très différents suggérait fortement pour ces structures une fonction digestive. D’où le nom de lysosomes qui leur fut attribué. Reposant originellement sur des critères biochimiques, le concept de lysosomes prit corps grâce à l’examen au microscope électronique de préparations purifiées. Dès 1956, une identification provisoire des lysosomes hépatiques aux entités décrites par les morphologistes sous le nom de corpuscules denses péri-canaliculaires [14] put être proposée, en collaboration avec Alex Novikoff, à qui l’on doit d’avoir ensuite utilisé sur une vaste échelle la technique de coloration cytochimique pour la phosphatase acide comme marqueur morphologique des lysosomes. L’identification des lysosomes fut définitivement confirmée en 1965, tandis que les peroxysomes, qui avaient eux aussi été purifiés, purent être reconnus comme correspondant aux «microcorpuscules» de la littérature [15].
Physiopathologie des lysosomes
Bien qu’il ait été proposé par des biochimistes, le concept de lysosome fut rapidement adopté par la communauté des cytologistes. Dès 1958, j’eus l’occasion de faire le point du sujet à une réunion tenue à Woods Hole [16]. Cinq ans plus tard, la Fondation Ciba à Londres servait d’hôte au premier symposium international sur les lysosomes, rassemblant tout un aréopage sous la présidence de James Danielli, le père du concept de la bi-couche lipidique. À cette époque, des lysosomes avaient déjà été identifiés dans plusieurs types cellulaires et on savait qu’ils étaient impliqués dans la digestion de matériel extracellulaire capté par endocytose et dans celle de matériel intracellulaire séquestré par autophagie. C’est lors de cette réunion, soit dit en passant, que furent créés les termes d’endocytose et d’autophagie, en même temps que celui d’exocytose. La participation possible des lysosomes dans certains processus pathologiques était déjà suspectée et même mise en évidence dans quelques cas. À partir de ce moment, le domaine ne fit que s’étendre, conduisant en 1969 à la publication d’une série d’ouvrages collaboratifs intitulée Lysosomes in Biology and Pathology [18] et publiée sous l’impulsion d’Honor B. Fell, une biologiste éminente connue pour la mise au point des cultures d’organes. Le septième et dernier volume de la série parut en 1984.
Parmi tous les travaux que je pourrais citer, je me contenterai d’en évoquer un seul, celui de Hers qui découvrit la première maladie lysosomiale, grâce, une fois de plus, à un heureux hasard. Dans des expériences réalisées dans un but fort différent, Hers fit la découverte imprévue qu’une a-glucosidase acide capable d’hydrolyser le glycogène était l’enzyme absente dans la glycogénose type II, maladie héréditaire grave caractérisée par des dépôts anormaux de glycogène dans de nombreux tissus [19]. Cette découverte ayant été faite dans un laboratoire où la notion d’hydrolase acide évoquait automatiquement celle de lysosomes, il ne fallut pas longtemps pour que l’enzyme en question fût reconnue comme appartenant à ces organites [20]. Devant ces données, Hers proposa alors que des molécules de glycogène, probablement intériorisées par autophagie, sont normalement digérées dans les lysosomes par la glucosidase et qu’elles ne le sont pas dans la maladie à cause de la déficience de cette enzyme. Cette hypothèse impliquait que les dépôts anormaux de glycogène devaient se trouver au sein de lysosomes, ce qui fut confirmé morphologiquement par la suite [21]. Par une brillante extrapolation, Hers généralisa cette hypothèse dans son concept de «maladie lysosomiale congénitale», définie comme un état pathologique où la déficience génétique d’une enzyme lysosomiale conduit à l’accumulation au sein des lysosomes de toutes les substances qui y sont normalement digérées par l’enzyme manquante [22]. Il existerait une maladie différente pour chaque enzyme. Venant à une époque où de nombreux biochimistes essayaient d’élucider les mécanismes pathogéniques responsables des mucopolysaccharidoses et des lipidoses, cette idée jeta une lumière unificatrice sur ce domaine très divers. Les spécialistes se rallièrent rapidement à la théorie lysosomiale. Six ans à peine après que cette théorie eût été formulée, de nombreux chercheurs s’unirent pour produire un impressionnant traité intitulé Lysosomes and Storage Diseases, publié en 1971 sous la direction de Géry Hers et de son collaborateur, François Van Hoof, malheureusement décédé en mai 2005 [23].
Conclusions
Rétrospectivement, trois conclusions se dégagent de ce bref rappel historique. Il y a d’abord le fait que les lysosomes (et les peroxysomes) ont été découverts par des biochimistes qui ont appliqué les méthodes classiques de leur discipline à des particules cellulaires. Ils ont commencé par caractériser des entités décelables biochimiquement en suivant une approche analytique, pour purifier ensuite ces entités par des moyens fondés sur les données analytiques et aboutir finalement à l’identification morphologique des entités purifiées. La plupart des cytologistes avaient, quant à eux, tendance à opter pour une démarche opposée, essayant d’abord de purifier des objets sur la base de critères morphologiques, pour en établir les propriétés biochimiques dans la suite. Les deux démarches ont leurs mérites et leurs avantages. Mais pour certains composants rares, tels que les lysosomes et les peroxysomes, l’approche morphologique se serait révélée extrêmement laborieuse, si tant est qu’elle eût pu réussir. Une leçon plus générale qu’on peut tirer de cette histoire est qu’il est souvent bénéfique de franchir les barrières interdisciplinaires.
Deuxième conclusion, il est d’importance capitale de comprendre les fondements physiques des techniques qu’on utilise. Dès le début, nous sommes retournés aux théories classiques de la centrifugation [24, 25] et nous les avons toujours appliquées dans nos travaux ultérieurs pour comprendre les techniques que nous utilisions, jauger leurs points forts et leurs faiblesses, en mettre au point de nouvelles et même, à l’occasion, construire de nouveaux instruments. Mes collaborateurs J. Berthet et H. Beaufay ont joué un rôle inestimable dans ces avancées [26, 27].
Enfin, je terminerai en insistant sur la valeur du hasard, la fée « serendipity », et, surtout, sur celle de son indispensable corollaire, la liberté de pouvoir suivre une voie nouvelle offerte par le hasard, quels que soient les engagements pris antérieurement. Il est remarquable que, dans la publication où les lysosomes sont mentionnés pour la première fois [1], les « Lilly Research Laboratories » soient remerciés pour leur soutien financier alors que leur principal sujet d’intérêt, l’insuline, en était notoirement absent. On ne peut que souhaiter que les organismes de financement d’aujourd’hui puissent faire preuve de la même largeur de vue.
Références
- de Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F. Biochem J 1955; 60 : 604–17. [Google Scholar]
- de Duve C. In: Dingle JT, Fell HB, eds. Lysosomes in biology and pathology, vol. 1. Amsterdam: North-Holland Publishing Company, 1969 : 3–40. [Google Scholar]
- de Duve C. J Biol Chem 2004; 279 : 21679–88. [Google Scholar]
- de Duve C, Berthet J, Hers HG, Dupret L. Bull Soc Chim Biol 1949; 31 : 1242–53. [Google Scholar]
- Claude A. J Exp Med 1946; 84 : 51–9. [Google Scholar]
- Claude A. J Exp Med 1946; 84 : 61–89. [Google Scholar]
- Hers HG, Berthet J, Berthet L, de Duve C. Bull Soc Chim Biol 1951; 33 : 21–41. [Google Scholar]
- Beaufay H, de Duve C. Bull Soc Chim Biol 1954; 36 : 1551–68. [Google Scholar]
- Berthet J, de Duve C. Biochem J 1951; 50 : 174–81. [Google Scholar]
- Berthet J, Berthet L, Appelmans F, de Duve C. Biochem J 1951; 50 : 182–9. [Google Scholar]
- Appelmans F, Wattiaux R, de Duve C. Biochem J 1955; 60 : 438–45. [Google Scholar]
- de Duve C, Berthet J, Berthet L, Appelmans F. Nature 1951; 167 : 389–92. [Google Scholar]
- de Duve C, Baudhuin P. Physiol Rev 1966; 46 : 323–57. [Google Scholar]
- Novikoff AB, Beaufay H, de Duve C. J Biophys Biochem Cytol 1956; 2 : 179–84. [Google Scholar]
- Baudhuin P, Beaufay H, de Duve C. J Cell Biol 1965; 26 : 219–43. [Google Scholar]
- de Duve C. In: Hayashi T, ed. Subcellular particles. New York: Ronald Press Company, 1958 : 128–59. [Google Scholar]
- de Reuck AVS, Cameron MP (eds). Ciba foundation symposium on lysosomes. London: J. and A. Churchill Ltd, 1963. [Google Scholar]
- Lysosomes in biology and pathology. Vol. 1. In: Dingle JT, Fell HB, eds. Amsterdam: North-Holland Publishing Company, 1969. Vol. 2. In: Dingle JT, Fell HB, eds. Amsterdam: North-Holland Publishing Company, 1969, 1973. Vol. 3. In: Dingle JT, ed. Amsterdam: North-Holland Publishing Company, 1973. Vol. 4. In: Dingle JT, Dean RT, eds. Amsterdam: North-Holland Publishing Company, 1975. Vol. 5. In: Dingle JT, Dean RT, eds. Amsterdam: North-Holland Publishing Company, 1976. Vol. 6. In: Dingle JT, Jacques PJ, Shaw IH, eds. Amsterdam: North-Holland Publishing Company, 1979. Vol. 7. In: Dingle JT, Dean RT, Sly W, eds. Amsterdam: Elsevier, 1984. [Google Scholar]
- Hers HG. Biochem J 1963; 86 : 11–6. [Google Scholar]
- Lejeune N, Thinès-Sempoux D, Hers HG. Biochem J 1963; 86 : 16–21. [Google Scholar]
- Baudhuin P, Hers HG, Loeb H. Lab Invest 1964; 13 : 1140–52. [Google Scholar]
- Hers HG. Gastroenterology 1965; 48 : 625–33. [Google Scholar]
- Hers HG, Van Hoof F (eds). Lysosomes and storage diseases. New York: Academic Press, 1973. [Google Scholar]
- de Duve C. The Harvey Lectures 1965; 59 : 49–87. [Google Scholar]
- de Duve C. Science 1975; 189 : 186–94. [Google Scholar]
- de Duve C, Berthet J. Int Rev Cytol 1954; 3 : 225–73. [Google Scholar]
- de Duve C, Berthet J, Beaufay H. Prog Biophys Biophys Chem 1959; 9 : 325–69. [Google Scholar]
Liste des figures
|
Figure 1. Lysosomes purifiés provenant de foie de rat (x10400). Reproduit du Journal of Cell Biology (1965; 26: 219-43), avec la permission de la Rockefeller University Press. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.