")
")
| Issue |
Med Sci (Paris)
Volume 42, Number 5, Mai 2026
Nos jeunes pousses ont du talent !
|
|
|---|---|---|
| Page(s) | 515 - 519 | |
| Section | Partenariat médecine/sciences - Écoles doctorales - Masters | |
| DOI | https://doi.org/10.1051/medsci/2026090 | |
| Published online | 29 mai 2026 | |
Séquençage d’ARN en cellule unique: Briser le mythe du clone
Single-cell RNA-sequencing: shattering the clone myth
1
Master 2 Microbiologie Fondamentale, Aix Marseille Université, Marseille, France
2
Aix Marseille Université, CNRS, LCB UMR7283, IMM, Marseille, France
a
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
b
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
c
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
d
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
e
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Dans le cadre de l’unité d’enseignement « Rédiger en sciences » d’Aix-Marseille Université, les étudiants du master 2 Microbiologie Intégrative et Fondamentale (MiF), en partenariat avec l’Institut de Microbiologie, Bioénergies et Biotechnologie (IM2B), ont été initiés aux exigences de l’écriture scientifique. Trois thématiques leur ont été proposées : les mécanismes d’adaptation au stress de l’enveloppe bactérienne, le séquençage d’ARN à l’échelle de la cellule unique et l´évolution de la pathogénicité des vibrios chez l´huître. À partir de publications originales, les étudiants ont rédigé des nouvelles scientifiques mettant en lumière les principaux résultats ainsi que la portée des travaux étudiés. Enrichi par des entretiens avec les auteurs, ces textes portent un regard original sur la compréhension du vivant dans les domaines de la microbiologie et de la santé.
Ces quatre auteurs ont contribué de façon égale au travail
© 2026 médecine/sciences – Inserm
 Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY (https://creativecommons.org/licenses/by/4.0), qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
L’actualité scientifique vue par les étudiants du Master 2 Microbiologie Intégrative et Fondamentale (MiF), en partenariat avec l’Institut de Microbiologie, Bioénergies et Biotechnologie (IM2B) d’Aix-Marseille Université

Responsable de l’Unité d’Enseignement
Laurent Aussel
Équipe pédagogique
Laurent Aussel – Professeur, Aix-Marseille Université – Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Sophie Tronnet – Professeure junior, Aix-Marseille Université – Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Sites web
https://sciences.univ-amu.fr/fr/formation/masters/master-microbiologie
Série coordonnée par Claire Deligne
À la fin du XIIIe siècle le terme « infection » désignait les souillures du péché dans un contexte spirituel, pour aboutir à la définition actuelle : la pénétration et la multiplication de germes pathogènes dans l’organisme [1]. Parallèlement à cette évolution conceptuelle est survenue la découverte des agents infectieux, les pathogènes [2]. Les techniques d’étude des bactéries n’ont depuis cessé de se perfectionner. De l’identification phénotypique rendue possible par le microscope optique au XVIIe siècle à des approches modernes d’identification génétique par séquençage développées à partir des années 2000, en passant par des méthodes enzymatiques rapides utilisées en laboratoire [3] (→).
(→) Voir m/s hors série n° 1, 2025, page 30
Malgré ces avancées, certains phénomènes demeurent mal compris, notamment la réponse adaptative hétérogène des bactéries à différents stress retrouvés en condition d’infection, provoqués par les défenses innées de l’hôte, la carence en nutriment ou encore l’exposition aux antibiotiques. C’est dans ce contexte qu’une variante du séquençage d’ARN (RNA-seq) a été développé pour l’étude bactérienne à l’échelle de la cellule unique.
L’hétérogénéité populationnelle façonne les infections
Dans des populations d’individus appartenant à une même lignée et évoluant dans un même environnement, des variations physiologiques dynamiques peuvent être observées. Cette hétérogénéité, dite phénotypique, résulte d’une diversité intrinsèque aux cellules. Elle se manifeste par une régulation différentielle ciblant des catégories de gènes : ceux impliqués dans le métabolisme, dans la réponse au stress ou encore de virulence. [4] Ces variations s’expriment aussi sous la forme de gradients d’expression pouvant être liés à l’exposition à différents microenvironnements (Figure 1A). Le bruit stochastique de l’expression génique (fluctuations aléatoires liées au faible nombre de molécules d’ARNm ou de protéines dans la cellule) et la distribution inégale de composants cellulaires lors de la division créent des états phénotypiques distincts. D’autres facteurs, tels que des oscillations périodiques (changements phénotypiques réguliers, par exemple le cycle cellulaire ou les horloges circadiennes), et les divisions asymétriques [4] contribuent à cette variabilité. Enfin, les interactions intercellulaires modulent les profils d’expression génique via des molécules de signalisation, diffusibles (comme dans le quorum sensing) ou ancrées à l’enveloppe. [4]
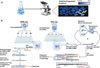 |
Figure 1 Description des deux méthodes de RNA-seq utilisées dans le cadre de l’étude de l’hétérogénéité phénotypique. A. Schéma d’observation d’une population clonale au microscope présentant une hétérogénéité phénotypique. B. Comparaison des méthodes de MATQ-seq et de PETRIseq. Le MATQ-seq est réalisé en plusieurs étapes : isolement des bactéries par cytométrie en flux, lyse des cellules, rétrotranscription de l’ARNm en ADNc avec un code barre unique, ajout d’une queue poly(C), amplification et séquençage. Le PETRI-seq est réalisé par perméabilisation des cellules, séparation dans différents puits, rétrotranscription de l’ARNm en ADNc, ajout d’un code barre unique associé au puits, regroupement des cellules et nouvelle séparation afin de répéter l’étape d’ajout du code barre deux fois. Les cellules sont enfin lysées pour préparer la librairie d’ADNc avant séquençage. |
Cette variabilité phénotypique influe sur la dynamique des infections. Elle permet à une fraction minoritaire de cellules d’adopter spontanément un état protecteur. Ce mécanisme, appelé bet-hedging, assure la survie d’une partie de la population face aux pressions de l’hôte, telles que la réponse immunitaire ou les traitements antibiotiques. Chez Escherichia coli, certaines sous-populations peuvent entrer dans un état de persistance : bien qu’elles restent viables, l’arrêt transitoire de leur division cellulaire confère une tolérance aux antibiotiques. Chez Salmonella enterica, une souspopulation ne produisant pas de flagelle (état fliC-OFF) échappe aux défenses de l’hôte [4]. Cette diversité favorise aussi une division du travail entre sous-populations. Lors d’une infection intestinale par S. enterica, la fraction exprimant le système de sécrétion de type III (ttss-1 ON) envahit l’épithélium et déclenche une réponse inflammatoire. En parallèle, la fraction ttss-1 OFF reste dans la lumière intestinale et profite des nutriments libérés [4]. Ces rôles complémentaires contribuent à la prolifération de la population bactérienne et à sa transmission. Les bactéries peuvent vivre sous forme de biofilms, qui correspondent à une communauté sessile enchâssée dans une matrice qu’elles sécrètent. La proximité cellulaire au sein de ces biofilms ou des tissus favorise la stabilité de ces interactions.
La coexistence de phénotypes cellulaires distincts au sein d’un même ensemble bactérien augmente les chances qu’une partie de la population résiste aux défenses de l’hôte et aux traitements. Cette hétérogénéité favorise également l’émergence de fonctions collectives, renforçant la virulence et facilitant la dissémination. Cette diversification, modulable par évolution, constitue un avantage pour les bactéries pathogènes. Son analyse nécessite d’explorer l’expression génique cellule par cellule, rendue possible par des approches de séquençage ARN en cellule unique (scRNA-seq).
Séquençage d’ARN en cellule unique
L’étude de la régulation de l’expression génique bactérienne a profondément évolué avec l’émergence du séquençage à haut débit. Cette avancée a permis une exploration globale du transcriptome grâce à la méthode de RNA-seq. Cette technique repose sur l’extraction des ARNm d’une population cellulaire, leur rétrotranscription en ADNc puis au séquençage des fragments obtenus (Figure 1B). L’analyse bioinformatique permet ensuite d’identifier et de quantifier les transcrits exprimés. Elle offre une vue d’ensemble de l’expression génique sans biais de sélection. Toutefois, cette approche fondée sur un mélange d’ARNm issus de nombreuses cellules masque l’hétérogénéité phénotypique. Afin de révéler cette hétérogénéité, des approches unicellulaires (scRNA-seq) ont été développées dans un premier temps chez les cellules eucaryotes. Cependant, ces techniques se reposaient sur la présence des queues polyadénylés des ARNm eucaryotes. Ainsi, des adaptations pour les modèles procaryotes ont vu le jour, dont le PETRI-seq et le MATQseq chez les bactéries.
Le PETRI-seq associe chaque ARN à sa cellule via des codes-barres, courtes séquences d’ADN uniques servant d’identifiants moléculaires. Les cellules sont fixées et perméabilisées au formaldéhyde, préservant leur intégrité tout en permettant la diffusion des réactifs. Elles sont ensuite réparties dans différents puits où s’effectue la rétrotranscription (Figure 1B). Lors de cette étape, un premier code-barres, propre à chaque puits, est ajouté à l’ADNc [5]. Les cellules sont ensuite regroupées, puis redistribuées dans de nouveaux puits. Deux cycles supplémentaires d’ajout de codebarres sont alors réalisés (Figure 1B). Au terme de ce processus, chaque molécule d’ADNc porte une combinaison unique de 3 codes-barres, dont la probabilité d’être partagée entre deux cellules est extrêmement faible [5]. Cette signature permet, lors du séquençage, d’attribuer les ARN à leur cellule d’origine et de reconstituer le profil transcriptomique individuel [6].
Le MATQ-seq constitue une autre approche de scRNA-seq reposant sur l’isolation physique de cellules individuelles. Elle utilise des dispositifs de microfluidique, tels que la cytométrie en flux, pour trier et distribuer chaque cellule dans un puits distinct. Une fois isolées, les cellules subissent les étapes classiques du RNA-seq couplées à l’ajout d’un code-barres spécifique à chaque puits (Figure 1B). Ce marquage permet de regrouper les transcrits issus d’une même cellule [7]. Cette stratégie permet de reconstruire les programmes transcriptionnels présents au sein de la population étudiée [7].
Ces deux approches diffèrent par leur complexité, leur coût et leur profondeur d’analyse : la MATQ-seq, plus coûteuse (tri cellulaire par cytométrie en flux), offre une plus grande profondeur de séquençage (300-600 gènes/cellule avec déplétion des rRNA), tandis que la PETRI-seq est plus économique et permet d’analyser plus de cellules. Toutes deux permettent néanmoins de révéler la diversité phénotypique des populations bactériennes soumises à des conditions identiques.
L’analyse de la cellule unique pour une meilleure compréhension de l’infection
La matrice des biofilms forme une structure dense, isolant et protégeant les bactéries du milieu extérieur. Grâce à cette protection, les bactéries du biofilm présentent une meilleure résistance aux différents stress environnementaux. Elles montrent également une sensibilité réduite aux antibactériens et autres composés extérieurs. Ainsi, les biofilms favorisent la survie des bactéries et compliquent l’identification des agents infectieux, rendant la prise en charge thérapeutique complexe. Ces communautés bactériennes structurées sont bien connues dans le domaine de la santé humaine. On les retrouve notamment comme constituants de la plaque dentaire, mais aussi adhérant à des dispositifs médicaux tels que les cathéters, les prothèses et les implants. L’hétérogénéité phénotypique est fortement présente dans les biofilms, influencée notamment par les interactions bactériennes, l’environnement extérieur du biofilm et son organisation spatiale [8].
Grâce au PETRI-seq, les chercheurs ont pu identifier dans un biofilm test, issue d’une culture in vitro d’E. coli, une sous-population représentant 2,1 % de l’ensemble de la culture, caractérisée par l’expression de seulement 10 gènes de l’enveloppe cellulaire. Ce groupe est constamment présent bien que son rôle n’ait pas encore été caractérisé [8]. Grâce à la finesse d’analyse de cette technique, il est à présent possible d’identifier des sous-populations peu abondantes. Ainsi, cette caractérisation de l’hétérogénéité au sein du biofilm permet d’envisager de nouvelles stratégies thérapeutiques ciblant différentes fractions de la population.
Un autre exemple d’application du scRNA-seq concerne le microbiote intestinal, dont l’équilibre est essentiel à la bonne santé humaine. Cette communauté microbienne compte plus de 10¹³ micro-organismes, dont une majorité de bactéries. Toutes sont exposées à des micro-environnements variables en raison d’une distribution inégale des nutriments dans le milieu. Cette hétérogénéité environnementale favorise l’émergence de phénotypes distincts. Parmi elles se trouve Bacteroides thetaiotaomicron, qui possède trois morphologies différentes [9]. Grâce au MATQ-seq, il a été montré que ces morphologies étaient directement liées à une spécialisation métabolique de la bactérie. Cette spécialisation influe sur la colonisation de l’hôte et la répartition dans la niche écologique [9]. Bien qu’aucune diversité fonctionnelle n’ait été clairement démontrée, l’importance du microbiote intestinal humain et l’abondance de cette espèce bactérienne dans celui-ci rendent ces résultats prometteurs. Ils ouvrent des perspectives pour la compréhension des dynamiques du microbiote intestinal, notamment les dysbioses.
Conclusion
Le scRNA-seq est aujourd’hui décliné en plusieurs méthodes chez les bactéries, comme le MATQ-seq, le PETRIseq ou encore BacDrop, une variante développée spécifiquement pour l’étude de l’hétérogénéité phénotypique en réponse à un stress induit par des antibiotiques [10].
L’existence de sous-populations hétérogènes est bien connue dans le contexte des infections, et plus largement comme un phénomène d’adaptation des bactéries aux environnements qu’elles rencontrent. Cependant, bien que leur étude soit possible au niveau de l’expression de gènes individuels grâce à des outils comme les rapporteurs transcriptionnels fluorescents, leur analyse à l’échelle du génome reste complexe. Dans ce cadre, les approches de scRNA-seq permettant d’étudier un grand nombre de cellules à l’échelle individuelle offrent des perspectives intéressantes. Néanmoins, leur développement récent ainsi que leur coût élevé, en particulier lié au séquençage, limitent encore leur utilisation à plus grande échelle et en contexte clinique.
Le scRNA-seq constitue ainsi un outil précieux, ouvrant la voie à une compréhension plus fine de la réponse des bactéries et au développement de nouvelles stratégies thérapeutiques.
Entretien avec Antoine-Emmanuel Saliba

© Helmholtz Institute
Antoine-Emmanuel Saliba est professeur à l’université de Wurtzbourg (Allemagne) et dirige le groupe Single-Cell Analysis au sein du Helmholtz Institute for RNA-based Infection Research (HIRI). Après un doctorat à l’Institut Curie, il s’est intéressé aux technologies permettant d’observer les cellules de manière individuelle. Aujourd’hui, avec son équipe, il cherche à comprendre pourquoi certaines bactéries persistent dans l’organisme malgré le système immunitaire et les traitements. Pour cela, il étudie les infections au sein des tissus, cellule par cellule, en combinant transcriptomique en cellule unique in vivo et analyses computationnelles avec l’objectif de relier organisation cellulaire et évolution de l’infection.
En quoi la mise en place du scRNA-seq chez les bactéries a été un défi comparé aux cellules eucaryotes ?
Chez les procaryotes, il y a trois grands problèmes qui se posent pour la mise en place du scRNA-seq. Dans un premier temps, la membrane bactérienne rend l’accès à l’ARN difficile sans traitement enzymatique préalable, ce qui n’est pas le cas pour les cellules eucaryotes. Ces membranes sont également propres à chaque espèce bactérienne, ce qui implique la mise en place de protocoles spécifiques à chaque bactérie. Cette variabilité complique le travail dans le cadre de l’étude d’associations de bactéries à l’échelle du microbiome par exemple où les protocoles utilisés vont devoir être efficaces sur toutes les bactéries analysées. De plus, chez les bactéries, l’ARN est présent en très faible quantité, de l’ordre du femtogrammes (10-15) contre des picogrammes (10-12) retrouvés chez les eucaryotes. Ce phénomène représente un défi majeur pour la sensibilité de détection des protocoles mis en œuvre. Enfin, chez les procaryotes, l’ARN n’est pas polyadénylé contrairement à l’ARN des eucaryotes. Cela signifie que lors de la capture des ARN, il va être impossible de différencier les ARN non codants, comme les ARN ribosomaux, des ARN messagers.
Comment le scRNA-seq a-t-il permis une compréhension et une caractérisation des bactéries sans biais expérimental ?
Le caractère non biaisé réside sur le principe même de la transcriptomique. En effet, cette approche ne sélectionne pas les transcrits capturés, ce qui permet une vision globale et impartiale de l’expression du génome. Le scRNA-seq est également une méthode qui tend à analyser le génome d’une cellule dans son entièreté malgré le fait qu’elle soit toujours limitée par certaines contraintes techniques telles que l’accessibilité à l’ARN et la sensibilité de détection.
Quelles applications thérapeutiques peut-on imaginer grâce aux scRNA-seq ?
J’ai trois exemples en tête : le premier concerne les populations bactériennes exposées à des antibiotiques. Dans cette population, le scRNA-seq va permettre de mettre en évidence les souspopulations qui présentent des phénotypes de résistance aux antibiotiques, ce qui permettra par la suite de pouvoir développer des stratégies thérapeutiques adaptées pour les cibler. Le second exemple se place dans le même contexte, mais concerne cette fois des phénotypes plus rares des bactéries : les persisters1. De la même manière, cette technique d’étude pourrait permettre de les identifier, de comprendre leur physiologie ainsi que leur adaptation à un environnement infectieux afin de concevoir des nouvelles stratégies pour empêcher leur développement. Le troisième exemple, plus complexe mais tout aussi intéressant, est celui du microbiote. Le scRNA-seq pourrait aider à mieux comprendre ses variations d’un point de vue phénotypique, en analysant l’expression des ARN bactériens dans différentes conditions, notamment en cas de maladie ou de dysbiose, et ce à l’échelle de la cellule unique, alors qu’à ce jour ces analyses ont uniquement été réalisées à l’échelle de la population.
Selon vous, comment la biologie actuelle se réinvente-t-elle face aux nouveaux outils et aux approches globales ?
Pour la première fois, il devient possible d’obtenir une description globale et systémique de toutes les molécules présentes dans la cellule. Grâce à cela, on peut désormais, sans réaliser aucune expérience, prévoir la réponse d’une cellule selon des modélisations et prédictions. Pour moi, c’est la véritable révolution que connaît actuellement la recherche, portée par une évolution extrêmement rapide des outils et des technologies.
Qu’est-ce qui vous a attiré vers le métier de chercheur et qu’est-ce qui vous a amené à votre poste actuel en Allemagne ?
Le premier point, c’est fondamentalement la curiosité : comprendre comment le vivant fonctionne et, d’un point de vue médical, comment les maladies infectieuses se développent et persistent. C’est un défi que nous rencontrons aujourd’hui et qui restera d’actualité pendant longtemps, avec l’émergence continue de nouvelles maladies infectieuses. Le deuxième point, c’est que j’apprécie particulièrement de développer de nouvelles technologies permettant de faire de la biologie quantitative. La recherche est un milieu incroyable qui nous offre cette possibilité, avec la liberté académique et le luxe de pouvoir mener des travaux de qualité.
Pour l’Allemagne, j’ai eu l’opportunité de participer à la création d’un nouvel institut, le Helmholtz Institute for RNA-based Infection Research (HIRI), dédié à la recherche sur l’ARN et les maladies infectieuses. C’était pour moi l’endroit idéal pour fonder mon laboratoire, une opportunité unique. Je pense aussi qu’en tant que chercheur, on a tout à gagner à avoir une réelle mobilité entre différentes cultures.
Au cours de vos travaux, vous avez étudié des organismes eucaryotes et procaryotes. Préférez-vous travailler sur un type d’organisme en particulier ou votre recherche se concentre-t-elle sur les aspects méthodologiques et l’amélioration des techniques, indépendamment du modèle biologique ?
Les deux aspects se combinent. Notre expertise reste centrée sur la réponse de l’hôte : il s’agit d’inventer de nouvelles méthodes pour mieux comprendre comment l’hôte réagit aux maladies infectieuses. Nous avons développé ces approches à différentes échelles, allant du micro-organisme unique jusqu’aux cohortes de patients. Le développement de nouvelles méthodes va toujours de pair avec la biologie et les organismes eux-mêmes : ils s’influencent mutuellement.
Si l’on veut étudier un processus infectieux à l’échelle de la cellule unique et qu’aucune méthode adaptée n’existe encore, il faut la créer. Cela pose de nombreux défis, à la fois technologiques et biologiques. On en a parlé pour les cellules procaryotes, mais il y a aussi la dimension temporelle : les processus infectieux se déroulent sur des échelles de temps très courtes, et il faut être capable de suivre les changements transcriptomiques sur ces périodes restreintes. Par ailleurs, il y a également la dimension spatiale : comment caractériser les réponses aux infections au sein de tissus complexes ? Tous ces aspects représentent des défis biologiques et technologiques importants, qui ne sont pas évidents à résoudre.
Selon vous, qu’est-ce qui fait aujourd’hui un bon chercheur : la créativité, la rigueur, ou la capacité à se remettre en question ?
Il y a les fondamentaux : être chercheur, c’est avant tout être capable de poser des questions et de créer des réseaux avec d’autres scientifiques. La curiosité et la motivation sont également des qualités essentielles dans ce métier. Mais il y a aussi un aspect plus difficile, souvent moins évident, qui est la capacité à encadrer et à gérer un laboratoire et une équipe, ce qui fait partie intégrante du métier de chercheur.
1Une bactérie dite persister voit son activité métabolique fortement diminuée, jusqu’à entrer en état de non-croissance, voir dormance. Cet état, rare, permet aux bactéries de fortement diminuer leur sensibilité aux antibiotiques.
Liens d’intérêt
Les auteurs déclarent ne pas avoir de lien d’intérêt
Références
- Infection : Définition de Infection. (s. d.). https://www.cnrtl.fr/definition/infection [Google Scholar]
- Freney J, Laurent F. Une brève histoire de la microbiologie. 1re ed. Paris : Eska, 2024. [Google Scholar]
- Tazi A. Maladies infectieuses en 2025. Populations vulnérables, défis diagnostiques et thérapeutiques. Med Sci (Paris) 2025 ; 41 (HS1) : 30-4. [Google Scholar]
- Ackermann M. A functional perspective on phenotypic heterogeneity in microorganisms. Nat Rev Microbiol 2015 ; 13 : 497-508. [Google Scholar]
- Blattman SB, Jiang W, Oikonomou P, et al. Prokaryotic single-cell RNA sequencing by in situ combinatorial indexing. Nat Microbiol 2020 ; 5 : 1192-201. [Google Scholar]
- Pountain AW, Jiang P, Yao T, et al. Transcription-replication interactions reveal bacterial genome regulation. Nature 2024 ; 626 : 661-9. [Google Scholar]
- Imdahl F, Vafadarnejad E, Homberger C, et al. Single-cell RNA-sequencing reports growth-condition-specific global transcriptomes of individual bacteria. Nat Microbiol 2020 ; 5 : 1202-6. [Google Scholar]
- Yan X, Liao H, Wang C, et al. An improved bacterial single-cell RNA-seq reveals biofilm heterogeneity. eLife 2024 ; 13 : RP97543. [Google Scholar]
- Bornet E, Prezza G, Cecchino L, et al. Low-input RNA-seq suggests metabolic specialization underlying morphological heterogeneity in a gut commensal bacterium. Cell Reports 2025 ; 44 : 115844. [Google Scholar]
- Ma P, Amemiya HM, He LL, et al. Bacterial droplet-based single-cell RNA-seq reveals antibiotic-associated heterogeneous cellular states. Cell 2024 ; 186 : 877-91. [Google Scholar]
Liste des figures
|
Figure 1 Description des deux méthodes de RNA-seq utilisées dans le cadre de l’étude de l’hétérogénéité phénotypique. A. Schéma d’observation d’une population clonale au microscope présentant une hétérogénéité phénotypique. B. Comparaison des méthodes de MATQ-seq et de PETRIseq. Le MATQ-seq est réalisé en plusieurs étapes : isolement des bactéries par cytométrie en flux, lyse des cellules, rétrotranscription de l’ARNm en ADNc avec un code barre unique, ajout d’une queue poly(C), amplification et séquençage. Le PETRI-seq est réalisé par perméabilisation des cellules, séparation dans différents puits, rétrotranscription de l’ARNm en ADNc, ajout d’un code barre unique associé au puits, regroupement des cellules et nouvelle séparation afin de répéter l’étape d’ajout du code barre deux fois. Les cellules sont enfin lysées pour préparer la librairie d’ADNc avant séquençage. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.