")
")
| Issue |
Med Sci (Paris)
Volume 31, Number 12, Décembre 2015
|
|
|---|---|---|
| Page(s) | 1083 - 1091 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20153112011 | |
| Published online | 16 décembre 2015 | |
Effets membranaires du récepteur alpha des œstrogènes
Une question de spécificité tissulaire
Tissue-specific membrane effects of estrogen receptor alpha
Inserm U1048-Université Paul Sabatier, I2MC, CHU Rangueil, 1, avenue Jean Poulhès, 31432 Toulouse, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
**
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les récepteurs des œstrogènes (ER) sont utilisés comme cibles thérapeutiques soit en les activant (par exemple dans le cas de la contraception ou de la ménopause), soit en les inhibant (comme pour le cancer du sein). Cependant, l’idéal serait de cibler chaque tissu spécifiquement. Certains modulateurs (les SERM, selective estrogen receptor modulator), spécifiques du récepteur alpha des œstrogènes (ERα), sont capables partiellement de réaliser ce ciblage spécifique. Il est donc important de comprendre la spécificité tissulaire de l’action de ERα afin d’optimiser le rapport bénéfice/risque de ces modulateurs. À côté de sa fonction nucléaire classique comme facteur de transcription, une fraction extranucléaire d’ERα relaye des actions rapides membranaires des œstrogènes. De nouveaux modèles de souris transgéniques ont récemment permis de dévoiler, dans chaque tissu, les rôles physiologiques et spécifiques des effets nucléaires et des effets membranaires d’ERα.
Abstract
Estrogen receptors (ER) are used as therapeutic targets, either for contraception or for the hormonal replacement therapy in menopausal women, but also in physiopathology for breast cancer treatment. It is therefore important to understand the tissue-specificity of the actions of ERα to optimize the benefits/risks ratio in each tissue. Besides the conventional nuclear ERα acting as a transcription factor, many studies have demonstrated that ERα is also able to mediate extra nuclear signaling, enabling rapid actions of estrogen. Recently, new transgenic mouse models were used to study these effects, and allowed to genetically segregate membrane versus nuclear actions of a steroid hormone receptor, demonstrating their in vivo tissue-specific roles.
© 2015 médecine/sciences – Inserm

Les récepteurs nucléaires interviennent, de manière spécifique pour chaque tissu, dans la transcription de gènes cibles impliqués dans la régulation de nombreuses fonctions physiologiques et pathologiques, comme le métabolisme des glucides et des lipides, l’inflammation, certains cancers et les maladies cardiovasculaires. Les récepteurs des œstrogènes (ER), ERα et ERβ, appartiennent à la superfamille des récepteurs nucléaires, facteurs de transcription dont le rôle est essentiel non seulement pour la reproduction féminine mais aussi masculine. ERα intervient également dans le maintien de l’homéostasie de nombreux tissus extra-reproducteurs. Le traitement hormonal de la ménopause et la contraception sont les principaux domaines médicaux qui s’intéressent à la modulation d’ERα. ERα est par ailleurs utilisé comme une cible majeure des thérapies adjuvantes anti-hormonales, pour les traitements des cancers du sein ERα positifs (ou hormono-dépendants), afin d’assurer un blocage de la croissance des cellules tumorales et prévenir une éventuelle récidive.
La synthèse endogène d’œstrogènes, et principalement de 17β-œstradiol (E2), diminue au moment de la ménopause (qui intervient en moyenne à l’âge de 51 ans). Ces hormones sexuelles peuvent donc être considérées comme n’étant pas essentielles après la vie reproductive. Cependant, cette chute des taux hormonaux conduit souvent à des troubles fonctionnels (syndrome climatérique affectant la qualité de vie) ainsi qu’à la perte de la protection vasculaire, métabolique et osseuse, démontrant un rôle important des œstrogènes dans ces tissus et ces fonctions extra-reproductrices. Un traitement hormonal substitutif (THS) basé sur la supplémentation en œstrogène associé à un progestatif, est utilisé pour prévenir le syndrome climatérique associé à la ménopause et pour conserver l’ensemble des effets protecteurs des œstrogènes. Cependant, l’étude Women Health Initiative (WHI)1 a mis un frein à l’utilisation de ce traitement en rapportant un risque accru de thrombose veineuse et de cancer du sein, et révélant un impact délétère sur les maladies coronariennes. Une nouvelle analyse de ces études nuance toutefois cette conclusion, en rapportant que le sur-risque coronarien n’apparaît que chez les femmes les plus âgées (ce qui est probablement dû à la prise du traitement après 20 ans de ménopause, et donc après une longue période de carence œstrogénique). Il semble donc que le mode d’administration du THS soit la clé de l’augmentation du risque de thrombose ; le type de progestatif utilisé apparaît aussi crucial pour le risque de cancer [1, 2]. Des modulateurs sélectifs des ER (SERM, selective estrogen receptor modulator)2 ont été développés dans le cadre du cancer du sein car ils conservent certaines actions bénéfiques (sur l’os par exemple) et s’opposent à certains effets délétères des œstrogènes, en particulier sur la prolifération du cancer du sein ER-positif [3]. Le SERM le plus utilisé est le tamoxifène, mais on a recours également au raloxifène.
Des mécanismes de résistance tumorale à ces traitements apparaissent dans environ la moitié des cas [4, 5]. La recherche de nouveaux SERM est par conséquent nécessaire pour le traitement de la ménopause, mais également pour le cancer du sein. Ceci requiert une meilleure compréhension du mode d’action de leur cible, le récepteur ERα.
Le récepteur alpha des œstrogènes (ERα) : un récepteur nucléaire
Le récepteur alpha des œstrogènes (ERα) a une activité nucléaire de facteur de transcription en régulant la transcription de gènes cibles en réponse au 17β-œstradiol (E2) [6]. Il comporte deux fonctions de transactivation : AF1 (activation function 1) située dans la région amino-terminale de la molécule, et AF2, dans sa région carboxy-terminale. Ces fonctions sont caractéristiques de la plupart des récepteurs nucléaires, et correspondent aux deux domaines actifs qui permettent le recrutement des protéines co-régulatrices modulant l’activité transcriptionnelle d’ERα. Il existe plusieurs isoformes d’ERα (de 66, 46 ou 36 kDa) qui présentent ou non les fonctions AF1 et/ou AF2 (Figure 1A). La fonction AF2 est composée de plusieurs hélices dont l’hélice 12, située dans le domaine de liaison du ligand (ligand binding domain, LBD) du récepteur. Le détail de cette structure, révélé par cristallographie aux rayons X, montre que la fixation de certains ligands modifie la conformation et le positionnement de cette hélice 12 et donc d’AF2, permettant alors les interactions avec des protéines de corégulation [7]. à l’inverse, la fonction AF1 est située dans le domaine amino-terminal de la protéine (Figure 1A), dont la structure, très labile, n’a pas pu encore être cristallographiée. Dans le cas des récepteurs des hormones stéroïdiennes, AF1 est souvent la fonction de transcription la plus active. Cependant, pour ERα, les deux domaines AF1 et AF2 peuvent entrer en synergie pour le recrutement de différents corégulateurs, selon un contexte qui diffère d’une cellule à l’autre et selon l’environnement du promoteur du gène cible [8–12]. Une interaction entre les domaines A (contenant la fonction AF1) et le domaine E (contenant la fonction AF2) de la protéine, a été mise en évidence suggérant qu’en réponse à la liaison de l’E2, le repositionnement de l’hélice 12 du domaine de fixation du ligand démasque et active non seulement la fonction AF2, mais aussi la fonction AF1, en libérant le domaine A de la protéine [13].
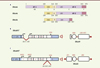 |
Figure 1. Isoformes d’ERα et construction des modèles murins mutés pour les fonctions AF1 ou AF2 de ERα. A. Structure des différentes isoformes du récepteur alpha des œstrogènes, ERα. DBD : DNA-binding domain ; H : hinge region ; LBD : ligand binding domain ; AF1, AF2 : fonctions de transactivation. B, C. Schématisation de la construction des modèles murins ERαAF10 et ERαAF20. AA : acides aminés ; AUG : codon d’initiation. |
Jusqu’à des études récentes, on pensait que seuls le type d’interaction et le niveau d’expression cellulaire des protéines corégulatrices (coactivateurs ou corépresseurs) des fonctions AF1 et AF2, déterminaient « l’ensemble récepteur-ligand-gène » conduisant soit à la stimulation, soit à l’inhibition, des effets biologiques tissu-spécifiques des œstrogènes [14]. La compréhension des spécificités cellulaires et tissulaires de ERα est d’une importance cruciale car elle conditionne l’optimisation de la synthèse et de l’utilisation des SERM. Cette question est cependant difficilement abordable in vitro avec des lignées cellulaires, et nécessite une approche intégrée in vivo. L’utilisation de souris transgéniques a récemment permis d’apporter une dissection moléculaire in vivo et un éclairage nouveau sur les mécanismes de spécificité cellulaire et tissulaire de ERα. Grâce à l’Institut clinique de la souris3 et de Pierre Chambon (Strasbourg, France), nous avons pu explorer le rôle et les fonctions des domaines AF1 et AF2 de ERa, en créant des souris transgéniques qui expriment une forme du récepteur ERα délétée spécifiquement, soit du domaine AB (souris ERαAF10), soit de sept acides aminés de l’hélice 12 du domaine AF2 (souris ERαAF20) (Figure 1B, C) [15, 16]. Dans le modèle ERαAF20, la grande majorité des effets bénéfiques de l’E2 sont abolis (athéroprotection, prévention du diabète de type 2 induit par une alimentation riche en graisse, déminéralisation osseuse) ainsi que ses effets délétères prolifératifs sur l’utérus [15, 17]. À l’inverse, dans le modèle de souris ERαAF10, ces effets bénéfiques sont conservés, démontrant que la fonction AF1 n’est pas nécessaire pour les effets bénéfiques du récepteur, mais les effets délétères prolifératifs sur l’utérus sont abolis [16, 18]. Ceci met donc en évidence une spécificité tissulaire dépendante des fonctions AF1 ou AF2, sur laquelle pourrait s’appuyer une stratégie de traitement (Figure 2A). Il est intéressant de noter que dans le modèle ERαAF10, qui présente une délétion du domaine AB, une isoforme tronquée de la protéine, de 49 kDa, proche de l’isoforme endogène de 46 kDa qui ne possède pas de fonction AF1, est générée (Figure 1B). Il apparaît donc que l’isoforme tronquée de 46 kDa est suffisante pour obtenir les effets vasculaires et métaboliques de l’E2 ; l’isoforme de pleine taille, de 66 kDa, est nécessaire pour les effets prolifératifs de l’hormone sur l’utérus (Figure 2A). De plus, la fonction AF2 est essentielle pour l’effet transcriptionnel de l’E2, à la fois sur l’utérus [18] et sur le foie (résultats non publiés), démontrant le rôle clé de cette fonction dans les effets nucléaires transcriptionnels de l’hormone. Korach et al. ont développé un autre modèle de souris altérée au niveau de ERαAF2 (AF2ERKI), en mutant les leucines 543 et 544 de l’hélice 12 en alanines [19]. Ce modèle a également révélé une hypoplasie utérine et des glandes mammaires rudimentaires. Ainsi, les deux modèles murins mutés sur la fonction AF2 présentent un phénotype similaire avec une absence de réponse transcriptionnelle à l’E2, faisant du modèle ERαAF20 un modèle d’inactivation des effets nucléaires d’ERα en réponse à l’E2.
 |
Figure 2. Mise en évidence des effets membranaires d’ERα au travers d’un modèle de ré-endothélialisation. A. Résumé de la dissection moléculaire du rôle des fonctions de transactivation AF1 et AF2 d’ERα in vivo. Le modèle de ré-endothélialisation apparaît en rouge, il met en évidence le rôle des effets membranaires in vivo. B. Illustration de l’effet de l’E2 dans le modèle de ré-endothélialisation : 3 jours après la lésion électrique de la carotide, le marquage au bleu Evans permet de visualiser la zone non cicatrisée ; l’E2 accélère la migration et la prolifération des cellules endothéliales composant l’intima (tunique interne d’un vaisseau sanguin). C. Conservation de l’effet de l’E2 sur la cicatrisation endothéliale dans les mutants nucléaires ERαAF10 et ERαAF20. WT : sauvage. |
Au cours de cette dissection moléculaire du rôle des fonctions AF1 et AF2 d’ERα, réalisée in vivo, nous avons également montré, dans un modèle de cicatrisation endothéliale de la carotide (Figure 2B), que l’accélération de la ré-endothélialisation (migration suivie de la prolifération de cellules endothéliales) induite par l’E2 est un processus qui ne dépend ni d’AF1, ni d’AF2 (ces effets de l’E2 étant perdus chez les souris ERαAF10 mais également chez les souris ERαAF20) [15, 17]. Pourtant, cet effet nécessite ERα mais pas ERβ (Figure 2 A, C) [49]. Cette observation surprenante nous a amenés à suspecter fortement l’implication d’une autre fonction d’ERα dans cet effet de protection endothéliale, et particulièrement celle de la fonction extranucléaire du récepteur.
Des effets rapides des œstrogènes aux effets membranaires : structure, conformation et signalisation du ERα membranaire
À côté de sa fonction nucléaire classique de facteur de transcription, de nombreuses études montrent qu’une fraction d’ERα est également localisée à la membrane de la cellule, et participe ainsi à une signalisation membranaire (appelée effets membranaires pour MISS : membrane initiated steroid signaling). Celle-ci a été initialement identifiée dans les actions rapides que présentent les œstrogènes (activation induite rapidement, de la seconde à plusieurs minutes). Dans des cellules endothéliales en culture, cette signalisation membranaire d’ERα, induite par E2, conduit à une activation aiguë de la monoxyde d’azote synthase endothéliale (endothelial nitric oxide synthase, eNOS) qui peut être inhibée à la fois par le ICI 182,780, un antagoniste de ER, et par le tamoxifène [20]. Un rôle potentiel d’ERβ a également été suggéré mais, par l’utilisation des modèles de souris transgéniques, nous avons pu clairement montrer que les deux principaux effets endothéliaux de l’E2 (correspondant à la stimulation de la production de monoxyde d’azote endothélial et la migration cellulaire) étaient médiés par ERα et non par ERβ [21, 22]. Si de nombreuses études se sont concentrées sur le rôle de ERα66, l’isoforme d’ERα de 66 kDa, il apparaît que l’isoforme tronquée ERα46 pourrait également être à l’origine des effets membranaires du récepteur [23, 24]. Comme évoqué précédemment, l’accélération de la cicatrisation endothéliale, induite par l’E2, persiste dans les modèles ERαAF10 qui n’expriment que la forme tronquée de 46/49 kDa du récepteur. Cette isoforme de la protéine est donc capable de médier les effets extranucléaires vasculaires attribués à ERα [15, 25]. L’isoforme ERα36, qui ne possède ni la fonction AF1, ni la fonction AF2, a été retrouvée à la membrane des cellules. Cette isoforme pourrait être impliquée dans la signalisation membranaire d’ERα, notamment, dans des cellules de cancer du sein, qui n’expriment aucune des autres isoformes du récepteur [26].
Le fractionnement des membranes plasmiques de cellules endothéliales a révélé une association de ERα extranucléaire avec en particulier, mais pas exclusivement, des cavéoles dans lesquelles le récepteur participe à une véritable plateforme de signalisation [20] (Figure 3). Une fraction d’ERα est en effet adressée à la membrane cytoplasmique grâce à la palmitoylation de la cystéine 447 qui permet son interaction avec la cavéoline-1 [27, 28] (Figure 3). Dans les artères ou les cellules endothéliales en culture, l’activation rapide de la eNOS (endothelial nitric oxide synthase) induite par l’E2 via ERα, implique plusieurs kinases : PI3K (phosphatidylinositol-3-kinase), AKT (protein kinase B), et ERK (extracellular-signal-regulated kinase) 1/2 [20] (Figure 3). Deux acteurs moléculaires apparaissent également être des partenaires directs d’ERα : 1) c-Src dont le domaine SH2 (Src homology 2) interagit avec la Tyr537 phosphorylée d’ERα [29, 30] et 2) p85a, la sous-unité régulatrice de PI3K. De manière intéressante, la méthylation transitoire d’ERα au niveau de l’Arg260 par l’arginine méthyltransférase PRMT1 (protein arginine methyltransferase 1), semble nécessaire pour que ces interactions aient lieu [31]. En effet, des mutations ponctuelles, localisées dans le domaine de liaison aux protéines G présent sur ERα (ERα-R256A, -D258A ou -R260A), ont permis pour la première fois d’obtenir des mutants ayant perdu les effets membranaires du récepteur in vitro (c’est-à-dire activation de la eNOS), mais conservant ses effets nucléaires (transcription génique), soulignant ainsi le rôle clé des résidus R256 et R258 dans la signalisation extranucléaire induite par ERα [32]. Outre l’activation de la eNOS au niveau de l’endothélium, des effets membranaires du récepteur ont également été décrits dans de nombreux autres types cellulaires. Ils induisent des voies de signalisation différentes comme les flux de calcium, la production d’AMPc et l’activation de nombreuses kinases (MEK [MAPK (mitogen-activated protein kinases)/ERK kinase], PI3K), impactant ainsi la prolifération et la migration cellulaire [20, 33]. Par ailleurs, cette signalisation d’origine membranaire peut également moduler l’activité transcriptionelle d’ERα via l’activation de kinases qui phosphorylent le récepteur nucléaire et modulent ainsi son activité nucléaire [34].
 |
Figure 3. Représentation schématique de l’adressage membranaire d’ERα. En parallèle de l’activité nucléaire d’ERα, ERα peut être palmitoylé et, ainsi, transporté à la membrane cytoplasmique, où il interagit avec la cavéoline. En présence d’E2, ERα va former une véritable plateforme de signalisation membranaire conduisant notamment à l’activation de l’eNOS (production de monoxyde d’azote [NO]) et à l’activation de nombreuses kinases impliquées dans des voies de prolifération et/ou de migration cellulaire. DHHC 7/21 : palmitoylacyltransférases 7/21. |
En ce qui concerne l’étude de la conformation de l’ERα membranaire, la topologie du récepteur associé avec la membrane plasmique n’en est encore qu’à ses balbutiements. Il a été montré qu’ERα, sous forme de monomère, active la eNOS [35], mais la présence de dimère du récepteur a également été mise en évidence à la membrane plasmique [36]. La question de la configuration du récepteur, au niveau de la membrane, reste donc une question ouverte. Les études menées par le groupe de Bender, consistant à faire exprimer à des cellules une forme de ERα46 marquée soit en amino-terminal, soit en carboxy-terminal, semblent démontrer que cette isoforme présente un ectodomaine en position carboxy-terminale [24]. Il est donc probable qu’il existe, là encore, plusieurs conformations membranaires possibles d’ERα, en fonction de ses partenaires protéiques. Un problème conceptuel soulevé par de nombreuses études est la nécessité d’utiliser des doses très importantes, supra-physiologiques, afin d’obtenir les effets membranaires de certains récepteurs stéroïdiens comme celui de la progestérone [37]. L’étude de l’affinité du récepteur ERα membranaire, a montré une diminution modérée de son affinité lorsqu’il était dans cette forme, surtout en ce qui concerne ERα46 [38]. Une autre approche possible pour aborder la question de la conformation membranaire d’ERα est l’utilisation des SERM (voir plus loin).
Approches pharmacologiques et génétiques pour étudier les effets membranaires d’ERa in vivo
Jusqu’à ces dernières années, notre capacité à distinguer rigoureusement les activités nucléaires et extranucléaires d’ERα était limitée. Concernant les approches pharmacologiques, l’E2 conjugué à l’albumine sérique bovine (afin d’empêcher son passage au travers de la membrane plasmique) a été utilisé initialement, principalement in vitro, mais sa stabilité est restée incertaine. Par la suite, J. Katzenellenbogen et al. ont synthétisé l’EDC (estrogen dendrimer conjugate), un œstrogène conjugué par une liaison covalente stable à un dendrimère afin d’empêcher sa pénétration dans le noyau cellulaire [39]. Lorsqu’il est administré in vivo, l’EDC n’active pas la transcription dépendante du ligand, comme en témoigne l’absence d’hypertrophie utérine et de régulation de l’expression génique dans l’utérus et le foie [40]. Cependant, comme l’E2, il est capable de stimuler in vitro, la prolifération et la migration de cellules endothéliales, et accélère la ré-endothélialisation, dans le modèle de cicatrisation endothéliale [40] (Figure 4). De manière très intéressante, l’activation par l’EDC des effets extranucléaires d’ERα, dont la voie impliquant ERK 1/2, ne stimule pas la croissance des cellules MCF-7, une lignée de cellules tumorales mammaires humaines, ni in vitro, ni in vivo (dans un modèle de xénogreffe chez la souris) [40]. L’EDC peut donc potentiellement être utilisé pour conférer une protection vasculaire, sans être à l’origine d’une augmentation du risque de cancer. Ainsi, l’EDC peut être considéré comme le premier SERM sélectif des effets extranucléaires, ou membranaires, d’ERα. Il permet l’activation des effets bénéfiques sur l’endothélium, mais n’induit pas la prolifération des cellules de cancer du sein.
 |
Figure 4. Mise en évidence de la spécificité tissulaire des effets membranaires et nucléaires d’ERα in vivo. Dans le modèle murin ERαC451A (A), la réponse utérine à l’E2 est conservée alors que les effets vasculaires sont abolis. À l’inverse, dans le modèle ErαAF20 (B), la réponse utérine à l’E2 est abolie alors que les effets vasculaires sont conservés (données originales publiées dans l’article [18]). WT : sauvage ; Veh : véhicule ; EDC : estrogen dendrimer conjugate. AA : acides aminés ; ns : non significatif ; les astérisques indiquent des différences significatives. |
Pour étudier in vivo, par des approches génétiques, les effets membranaires d’ERα, il était d’abord nécessaire de comprendre le mécanisme d’adressage et d’ancrage du récepteur à la membrane plasmique de la cellule. Un site de palmitoylation, essentiel à la localisation membranaire de ERα, a été identifié au niveau de la Cys-447 d’ERα, qui est située dans le domaine de liaison du ligand [27, 41]. Sur la base de ces travaux, réalisés in vitro, nous avons donc généré un modèle de souris présentant une mutation ponctuelle au niveau de ce site de palmitoylation (C451AERα, Figure 4A) [18]. L’altération de la localisation membranaire d’ERα ainsi induite, a été confirmée in vivo, dans des hépatocytes primaires. La souris femelle présentant la mutation C451AERα est totalement stérile. Cette infertilité est associée à une dysfonction ovarienne. Cependant, l’action de l’E2 sur l’utérus de ces souris est entièrement préservée : la régulation de l’expression génique (à l’exception de quelques gènes) et la prolifération de l’épithélium luminal de l’endomètre, sont en effet relativement similaires chez les souris mutantes C451AERα et chez les souris de type sauvage. En revanche, les effets vasculaires de l’E2, comme l’activation de la eNOS, la vasorelaxation de l’artère mésentérique et l’accélération de la ré-endothélialisation, sont abrogés chez les souris mutantes (Figure 4A) [18]. Le modèle C451AERα est donc le premier modèle de perte de fonction spécifique de l’activité membranaire d’ERα. E. Levin a également généré un modèle de souris, nommé NOER (pour nuclear only ER), portant la même mutation ponctuelle d’ERα (C451A). Ces souris présentent un phénotype similaire aux souris C451AERα, en ce qui concerne la fertilité, mais une différence notable, en ce qui concerne le phénotype utérin [42]. Cette divergence, qui reste intrigante, pourrait résulter des stratégies utilisées pour la construction des deux modèles. Le modèle d’absence d’effets nucléaires ERαAF20 que nous avons décrit précédemment, est complémentaire du modèle C451AERα pour étudier les rôles respectifs des effets membranaires et nucléaires de ERα. En effet, dans ce modèle, on observe que l’accélération de la ré-endothélialisation en réponse à l’E2, ou à l’EDC, est conservée (Figure 4B) alors que l’analyse à large échelle de l’expression génique en réponse à l’E2, au niveau de l’utérus, montre une perte presque totale des effets transcriptionnels en réponse à l’E2. Dans ce modèle ERαAF20, les effets dits membranaires de l’E2 sont donc préservés tandis que ses effets nucléaires sont abolis. À l’inverse, dans le modèle C451A-ERα, la régulation de l’expression génique dans l’utérus est presque totalement conservée, alors que les effets membranaires vasculaires sont abolis [18]. Ces études ont donc permis, pour la première fois, de montrer le rôle physiologique d’ERα lorsqu’il est localisé à la membrane, mais aussi de différencier, par une approche de génétique, les effets nucléaires et membranaires d’un récepteur aux hormones stéroïdiennes, démontrant ainsi in vivo leur spécificité tissulaire (Figure 4).
Vers une nouvelle vision de la spécificité tissulaire des SERM
Les résultats que nous avons présentés ici semblent réfuter l’hypothèse selon laquelle la spécificité tissulaire des SERM s’expliquerait uniquement par le recrutement par l’ERα nucléaire de panels différents de coactivateurs ou de corépresseurs [14, 43]. Pour les SERM déjà connus, il a été montré, in vitro, que l’ICI 182,780 et le tamoxifène avaient une activité antagoniste sur l’induction de la eNOS [20]. In vivo, nous avons récemment confirmé que le tamoxifène n’accélère pas la ré-endothélialisation [44] mais qu’il antagonise l’effet de l’E2 sur ce processus membranaire (non publié). Il apparaît donc, à présent, que la spécificité tissulaire des SERM ne dépend pas seulement du recrutement des cofacteurs sur AF1 et AF2 (effets nucléaires), mais aussi d’une signalisation membranaire d’ERα. Cette spécificité peut désormais être étudiée en combinant l’utilisation des nouveaux modèles de souris transgéniques développés et des outils pharmacologiques sélectifs. L’EDC, composé synthétique d’une part, et l’estétrol (E4), un œstrogène naturel produit uniquement pendant la grossesse d’autre part, semblent à cet égard tout à fait intéressants.
L’EDC qui, comme nous l’avons déjà évoqué, a permis l’étude in vivo des effets membranaires du récepteur, a également permis d’évaluer in vitro, l’impact de l’activation du ERα membranaire, sur la régulation de l’expression génique des cellules du cancer du sein MCF-7 [45]. L’analyse à large échelle du génome, par biopuces (microarray), des cellules stimulées, a conduit à l’identification, parmi les gènes induits par E2, d’un sous-ensemble de gènes (25 %) également sensibles à l’EDC. Le traitement par des antagonistes de ERα, ou son altération par la technique de siRNA (small interfering RNA), abolissent la stimulation, par l’EDC, de l’expression génique indiquant que ERα est à l’origine de cet effet membranaire sur la transcription. De plus, les inhibiteurs de MAP kinases et de c-Src suppriment la stimulation de l’expression génique induite par l’E2 et par l’EDC. L’analyse par immunoprécipitation de la chromatine, a révélé que l’EDC, contrairement à l’E2, est incapable de recruter ERα au niveau des séquences régulatrices des gènes cibles. Ces résultats suggèrent que, par l’activation de kinases ou autres signaux, l’EDC initie des modifications au niveau des facteurs de transcription qui sont recrutés sur les régions régulatrices des gènes régulés [45].
L’estétrol (E4) est un œstrogène produit uniquement par le foie fœtal durant la grossesse chez l’homme [46]. L’E4 a été découvert il y a plusieurs décennies, mais, considéré comme un œstrogène « faible » (c’est-à-dire de faible activité), il a longtemps été ignoré. En effet, bien que l’E4 ait une affinité 100 fois plus faible pour ERα que l’E2, nous avons montré que des doses élevées de cet œstrogène étaient capables d’induire les effets nucléaires d’ERα, conduisant à la prolifération épithéliale dans l’utérus et à la prévention de l’athérome dans des souris hypercholestérolémiques, et ceci de manière similaire à ce qu’on observe avec l’E2 [47]. Cependant, contrairement à l’E2, et avec les doses utilisées, l’E4 n’induit pas les effets membranaires du récepteur comme la libération de monoxyde d’azote endothélial et l’accélération de la ré-endothélialisation (Figure 5). En outre, l’E4 inhibe les effets membranaires de l’E2 sur l’endothélium, mais également sur des lignées de cancer du sein. L’E4 apparaît donc comme le premier et probablement le seul SERM endogène, capable d’activer les effets nucléaires d’ERα mais qui est dépourvu des effets membranaires du récepteur, pouvant même s’y opposer, en combinaison avec l’E2 [47].
 |
Figure 5. Schématisation de la spécificité tissulaire des SERM (selective estrogen receptor modulator). Exemples de l’E4 (Estetrol), qui est un faible agoniste de l’activité nucléaire et un antagoniste de l’activité membranaire d’ERα, et de l’EDC (estrogen dendrimer conjugate), qui active uniquement les effets membranaires d’ERα. CE : cellules endothéliales ; EP : cellules épithéliales. |
Ainsi, l’EDC et l’E4 apparaissent comme deux outils pharmacologiques complémentaires. Il semble que l’E4 puisse présenter un intérêt médical : dans un essai clinique de phase II, il a été évalué en combinaison avec un progestatif, comme nouveau contraceptif oral. Il inhibe le pic d’hormone lutéinisante (LH), et donc l’ovulation, mais ne provoque pas de changement dans la synthèse des facteurs hépatiques circulants (C. Kluft Cornelis et al., manuscrit en préparation) [50]. Il pourrait ainsi ne pas influer sur les événements participant à la thrombose veineuse, contrairement aux produits pharmaceutiques comprenant de l’E2 ou de l’éthinylœstradiol [48]. Des données préliminaires indiquent clairement que dans le foie, la régulation de l’expression des gènes par ERα repose à la fois sur les effets nucléaires et membranaires du récepteur. Le découplage des effets membranaires et nucléaires par l’E4 pourrait donc, selon notre hypothèse, permettre d’atténuer les effets hépatiques des œstrogènes et donc présenter un profil liver friendly 4, caractéristique unique pour un SERM.
Conclusion
ERα, considéré jusqu’à récemment uniquement comme un récepteur nucléaire, peut présenter deux localisations subcellulaires qui lui permettent d’assurer les nombreuses fonctions physiologiques dans lesquelles il est impliqué. La répartition de ces localisations, nucléaires et/ou membranaires, pourrait être spécifique du tissu. Les effets physiologiques de ces pools de récepteurs résultent cependant d’une dynamique complexe entre les deux formes. Les mécanismes de régulation conduisant à l’une ou l’autre des deux localisations, leurs interactions et l’implication des différentes isoformes du récepteur, restent des questions ouvertes. Les réponses à ces questions dépendent probablement du type, et même de l’état de différenciation, de la cellule. Il s’agit donc, à présent, d’étudier cette dynamique entre les différentes localisations de ERα, afin de mieux comprendre la spécificité d’action des œstrogènes, et ainsi d’optimiser la modulation du récepteur ERα dans le cadre du traitement de la ménopause et du cancer du sein.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
L’initiative sur la santé des femmes (WHI) est une étude à long terme axée sur les stratégies de prévention des maladies cardiaques, des cancers du sein et colorectal et des fractures ostéoporotiques chez les femmes ménopausées. Commencée en 1993, elle compte 161 808 femmes âgées de 50 à 79 ans.
Les SERM sont des molécules capables de se fixer sur les récepteurs des œstrogènes et d’avoir une action à la fois agoniste et antagoniste des œstrogènes.
L’Institut Clinique de la Souris (ICS) est une infrastructure spécialisée dans la recherche translationnelle et la génomique fonctionnelle.
Ami du foie.
Références
- Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007 ; 297 : 1465–1477. [CrossRef] [PubMed] [Google Scholar]
- Lenfant F, Tremollieres F, Gourdy P, Arnal JF. Timing of the vascular actions of estrogens in experimental and human studies: why protective early, and not when delayed? Maturitas 2011 ; 68 : 165–173. [CrossRef] [PubMed] [Google Scholar]
- McDonnell DP, Wardell SE. The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr Opin Pharmacol 2010 ; 10 : 620–628. [CrossRef] [PubMed] [Google Scholar]
- Palmieri C, Patten DK, Januszewski A, et al. Breast cancer: current and future endocrine therapies. Mol Cell Endocrinol 2014 ; 382 : 695–723. [CrossRef] [PubMed] [Google Scholar]
- Jiang Q, Zheng S, Wang G. Development of new estrogen receptor-targeting therapeutic agents for tamoxifen-resistant breast cancer. Future Med Chem 2013 ; 5 : 1023–1035. [CrossRef] [PubMed] [Google Scholar]
- Heldring N, Pike A, Andersson S, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev 2007 ; 87 : 905–931. [CrossRef] [PubMed] [Google Scholar]
- Lonard DM, O’Malley BW. Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol 2012 ; 8 : 598–604. [CrossRef] [PubMed] [Google Scholar]
- Kobayashi Y, Kitamoto T, Masuhiro Y, et al. p300 mediates functional synergism between AF-1 and AF-2 of estrogen receptor alpha and beta by interacting directly with the N-terminal A/B domains. J Biol Chem 2000 ; 275 : 15645–15651. [CrossRef] [PubMed] [Google Scholar]
- Metivier R, Penot G, Flouriot G, Pakdel F. Synergism between ERalpha transactivation function 1 (AF-1) and AF-2 mediated by steroid receptor coactivator protein-1: requirement for the AF-1 alpha-helical core and for a direct interaction between the N- and C-terminal domains. Mol Endocrinol 2001 ; 15 : 1953–1970. [PubMed] [Google Scholar]
- Onate SA, Boonyaratanakornkit V, Spencer TE, et al. The steroid receptor coactivator-1 contains multiple receptor interacting and activation domains that cooperatively enhance the activation function 1 (AF1) and AF2 domains of steroid receptors. J Biol Chem 1998 ; 273 : 12101–12108. [CrossRef] [PubMed] [Google Scholar]
- Benecke A, Chambon P, Gronemeyer H. Synergy between estrogen receptor α activation functions AF1 and AF2 mediated by transcription intermediary factor TIF2. EMBO Rep 2000 ; 1 : 151–157. [CrossRef] [PubMed] [Google Scholar]
- Tzukerman MT, Esty A, Santiso-Mere D, et al. Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol Endocrinol 1994 ; 8 : 21–30. [PubMed] [Google Scholar]
- Metivier R, Stark A, Flouriot G, et al. A dynamic structural model for estrogen receptor-alpha activation by ligands, emphasizing the role of interactions between distant A and E domains. Mol Cell 2002 ; 10 : 1019–1032. [CrossRef] [PubMed] [Google Scholar]
- Katzenellenbogen BS, Katzenellenbogen JA. Biomedicine. Defining the S in SERMs. Science 2002 ; 295 : 2380–2381. [Google Scholar]
- Billon-Gales A, Krust A, Fontaine C, et al. Activation function 2 (AF2) of estrogen receptor-[alpha] is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc Natl Acad Sci USA 2011 ; 108 : 13311–13316. [CrossRef] [Google Scholar]
- Billon-Gales A, Fontaine C, Filipe C, et al. The transactivating function 1 of estrogen receptor[alpha] is dispensable for the vasculoprotective actions of 17[beta]-estradiol. Proc Natl Acad Sci USA 2009 ; 106 : 2053–2058. [CrossRef] [Google Scholar]
- Adlanmerini M, Solinhac R, Abot A, et al. Mutation of the palmitoylation site of estrogen receptor alpha in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci USA 2014 ; 111 : E283–E290. [CrossRef] [Google Scholar]
- Handgraaf S, Riant E, Fabre A, et al. Prevention of obesity and insulin resistance by estrogens requires ERalpha activation function-2 (ERalphaAF-2), whereas ERalphaAF-1 is dispensable. Diabetes 2013 ; 62 : 4098–4108. [CrossRef] [PubMed] [Google Scholar]
- Arao Y, Hamilton KJ, Ray MK, et al. Estrogen receptor alpha AF-2 mutation results in antagonist reversal and reveals tissue selective function of estrogen receptor modulators. Proc Natl Acad Sci USA 2011 ; 108 : 14986–14991. [CrossRef] [Google Scholar]
- Wu Q, Chambliss K, Umetani M, et al. Non-nuclear estrogen receptor signaling in the endothelium. J Biol Chem 2011 ; 286 : 14737–14743. [CrossRef] [PubMed] [Google Scholar]
- Brouchet L, Krust A, Dupont S, et al. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-alpha but not estrogen receptor-beta. Circulation 2001 ; 103 : 423–428. [CrossRef] [PubMed] [Google Scholar]
- Darblade B, Pendaries C, Krust A, et al. Estradiol alters nitric oxide production in the mouse aorta through the alpha-, but not beta-, estrogen receptor. Circ Res 2002 ; 90 : 413–419. [CrossRef] [Google Scholar]
- Russell KS, Haynes MP, Sinha D, et al. Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc Natl Acad Sci USA 2000 ; 97 : 5930–5935. [CrossRef] [Google Scholar]
- Kim KH, Toomre D, Bender JR. Splice isoform estrogen receptors as integral transmembrane proteins. Mol Biol Cell 2011 ; 22 : 4415–4423. [CrossRef] [PubMed] [Google Scholar]
- Kim KH, Bender JR. Membrane-initiated actions of estrogen on the endothelium. Mol Cell Endocrinol 2009 ; 308 : 3–8. [CrossRef] [PubMed] [Google Scholar]
- Chaudhri RA, Hadadi A, Lobachev KS, et al. Estrogen receptor-alpha 36 mediates the anti-apoptotic effect of estradiol in triple negative breast cancer cells via a membrane-associated mechanism. Biochim Biophys Acta 2014 ; 1843 : 2796–2806. [CrossRef] [PubMed] [Google Scholar]
- Acconcia F, Ascenzi P, Bocedi A, et al. Palmitoylation-dependent estrogen receptor alpha membrane localization: regulation by 17beta-estradiol. Mol Biol Cell 2005 ; 16 : 231–237. [CrossRef] [PubMed] [Google Scholar]
- Acconcia F, Ascenzi P, Fabozzi G, et al. S-palmitoylation modulates human estrogen receptor-alpha functions. Biochem Biophys Res Commun 2004 ; 316 : 878–883. [CrossRef] [PubMed] [Google Scholar]
- Migliaccio A, Castoria G, Di Domenico M, et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J 2000 ; 19 : 5406–5417. [CrossRef] [PubMed] [Google Scholar]
- Li L, Hisamoto K, Kim KH, et al. Variant estrogen receptor-c-Src molecular interdependence and c-Src structural requirements for endothelial NO synthase activation. Proc Natl Acad Sci USA 2007 ; 104 : 16468–16473. [CrossRef] [Google Scholar]
- Le Romancer M, Treilleux I, Leconte N, et al. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell 2008 ; 31 : 212–221. [CrossRef] [PubMed] [Google Scholar]
- Wu Q, Chambliss K, Lee WR, et al. Point mutations in the ERalpha Galphai binding domain segregate nonnuclear from nuclear receptor function. Mol Endocrinol 2013 ; 27 : 2–11. [CrossRef] [PubMed] [Google Scholar]
- Marino M, Ascenzi P. Membrane association of estrogen receptor alpha and beta influences 17beta-estradiol-mediated cancer cell proliferation. Steroids 2008 ; 73 : 853–858. [CrossRef] [PubMed] [Google Scholar]
- La Rosa P, Pesiri V, Leclercq G, et al. Palmitoylation regulates 17beta-estradiol-induced estrogen receptor-alpha degradation and transcriptional activity. Mol Endocrinol 2012 ; 26 : 762–774. [CrossRef] [PubMed] [Google Scholar]
- Chambliss KL, Simon L, Yuhanna IS, et al. Dissecting the basis of nongenomic activation of endothelial nitric oxide synthase by estradiol: role of ERalpha domains with known nuclear functions. Mol Endocrinol 2005 ; 19 : 277–289. [CrossRef] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Merchenthaler I, et al. Plasma membrane estrogen receptors exist and functions as dimers. Mol Endocrinol 2004 ; 18 : 2854–2865. [CrossRef] [PubMed] [Google Scholar]
- Wendler A, Baldi E, Harvey BJ, et al. Position paper: rapid responses to steroids. Current status and future prospects. Eur J Endocrinol 2010 ; 162 : 825–830. [CrossRef] [PubMed] [Google Scholar]
- Lin AHY, Li RWS, Ho EYW, et al. Differential ligand binding affinities of human estrogen receptor-α isoforms. PLoS One 2013 ; 8 : e63199. [CrossRef] [PubMed] [Google Scholar]
- Harrington WR, Kim SH, Funk CC, et al. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol 2006 ; 20 : 491–502. [CrossRef] [PubMed] [Google Scholar]
- Chambliss KL, Wu Q, Oltmann S, et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest 2010 ; 120 : 2319–2330. [CrossRef] [PubMed] [Google Scholar]
- Li L, Haynes MP, Bender JR. Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proc Natl Acad Sci USA 2003 ; 100 : 4807–4812. [CrossRef] [Google Scholar]
- Pedram A, Razandi M, Lewis M, et al. Membrane-localized estrogen receptor a is required for normal organ development and function. Dev Cell 2014 ; 29 : 482–490. [CrossRef] [PubMed] [Google Scholar]
- Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev 2004 ; 25 : 45–71. [CrossRef] [PubMed] [Google Scholar]
- Fontaine C, Abot A, Billon-Gales A, et al. Tamoxifen elicits atheroprotection through estrogen receptor alpha AF-1 but does not accelerate reendothelialization. Am J Pathol 2013 ; 183 : 304–312. [CrossRef] [PubMed] [Google Scholar]
- Madak-Erdogan Z, Kieser KJ, Kim SH, et al. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol 2008 ; 22 : 2116–2127. [CrossRef] [PubMed] [Google Scholar]
- Visser M, Foidart JM. Coelingh Bennink HJ. In vitro effects of estetrol on receptor binding, drug targets and human liver cell metabolism. Climacteric 2008 ; 11 (suppl 1) : 64–68. [CrossRef] [PubMed] [Google Scholar]
- Abot A, Fontaine C, Buscato M, et al. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor alpha modulation, uncoupling nuclear and membrane activation. EMBO Mol Med 2014 ; 10 : 1328–1346. [CrossRef] [PubMed] [Google Scholar]
- Canonico M, Plu-Bureau G, Lowe GD, Scarabin PY. Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: systematic review and meta-analysis. BMJ 2008 ; 336 : 1227–1231. [CrossRef] [PubMed] [Google Scholar]
- Brouchet L, Krust A, Dupont S, et al. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-alpha but not estrogen receptor-beta. Circulation 2001 ; 103 : 423–428. [CrossRef] [PubMed] [Google Scholar]
- Mawet M, Maillard C, Klipping C, et al. Unique effects on hepatic function, lipid metabolism, bone and growth endocrine parameters of estetrol in combined oral contraceptives. Eur J Contracept Reprod Health Care 2015 ; 20 : 463–475. [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Isoformes d’ERα et construction des modèles murins mutés pour les fonctions AF1 ou AF2 de ERα. A. Structure des différentes isoformes du récepteur alpha des œstrogènes, ERα. DBD : DNA-binding domain ; H : hinge region ; LBD : ligand binding domain ; AF1, AF2 : fonctions de transactivation. B, C. Schématisation de la construction des modèles murins ERαAF10 et ERαAF20. AA : acides aminés ; AUG : codon d’initiation. |
| Dans le texte | |
|
Figure 2. Mise en évidence des effets membranaires d’ERα au travers d’un modèle de ré-endothélialisation. A. Résumé de la dissection moléculaire du rôle des fonctions de transactivation AF1 et AF2 d’ERα in vivo. Le modèle de ré-endothélialisation apparaît en rouge, il met en évidence le rôle des effets membranaires in vivo. B. Illustration de l’effet de l’E2 dans le modèle de ré-endothélialisation : 3 jours après la lésion électrique de la carotide, le marquage au bleu Evans permet de visualiser la zone non cicatrisée ; l’E2 accélère la migration et la prolifération des cellules endothéliales composant l’intima (tunique interne d’un vaisseau sanguin). C. Conservation de l’effet de l’E2 sur la cicatrisation endothéliale dans les mutants nucléaires ERαAF10 et ERαAF20. WT : sauvage. |
| Dans le texte | |
|
Figure 3. Représentation schématique de l’adressage membranaire d’ERα. En parallèle de l’activité nucléaire d’ERα, ERα peut être palmitoylé et, ainsi, transporté à la membrane cytoplasmique, où il interagit avec la cavéoline. En présence d’E2, ERα va former une véritable plateforme de signalisation membranaire conduisant notamment à l’activation de l’eNOS (production de monoxyde d’azote [NO]) et à l’activation de nombreuses kinases impliquées dans des voies de prolifération et/ou de migration cellulaire. DHHC 7/21 : palmitoylacyltransférases 7/21. |
| Dans le texte | |
|
Figure 4. Mise en évidence de la spécificité tissulaire des effets membranaires et nucléaires d’ERα in vivo. Dans le modèle murin ERαC451A (A), la réponse utérine à l’E2 est conservée alors que les effets vasculaires sont abolis. À l’inverse, dans le modèle ErαAF20 (B), la réponse utérine à l’E2 est abolie alors que les effets vasculaires sont conservés (données originales publiées dans l’article [18]). WT : sauvage ; Veh : véhicule ; EDC : estrogen dendrimer conjugate. AA : acides aminés ; ns : non significatif ; les astérisques indiquent des différences significatives. |
| Dans le texte | |
|
Figure 5. Schématisation de la spécificité tissulaire des SERM (selective estrogen receptor modulator). Exemples de l’E4 (Estetrol), qui est un faible agoniste de l’activité nucléaire et un antagoniste de l’activité membranaire d’ERα, et de l’EDC (estrogen dendrimer conjugate), qui active uniquement les effets membranaires d’ERα. CE : cellules endothéliales ; EP : cellules épithéliales. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.