")
")
| Issue |
Med Sci (Paris)
Volume 31, Number 2, Février 2015
|
|
|---|---|---|
| Page(s) | 180 - 186 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20153102015 | |
| Published online | 4 mars 2015 | |
PCR digitale en micro-compartiments
II. Apport pour la détection quantitative d’ADN tumoral circulant
Digital PCR compartmentalization II. Contribution for the quantitative detection of circulating tumor DNA
1
Université Paris Sorbonne Cité, Inserm UMR-S1147, 45, rue des Saints-Pères, 75270, Paris, France
2
Département de biologie, hôpital européen Georges Pompidou, AP-HP, Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La recherche d’altérations génétiques dans les cellules tumorales fait partie de la prise en charge des patients atteints de cancer. L’analyse de l’ADN libre tumoral circulant dans les fluides biologiques, dont le sang, pourrait permettre le dépistage précoce de la maladie, la gestion thérapeutique des patients et la détection de récidives. Cet ADN libre tumoral, décrit comme représentatif de l’hétérogénéité tumorale, peut être retrouvé dans le sang dans des proportions faibles et variables selon la nature et la progression de la tumeur. Son analyse nécessite des méthodes à la fois très sensibles, spécifiques et quantitatives. Ces exigences représentaient une limite technologique que les récentes avancées de la PCR (polymerase chain reaction) digitale devraient permettre de dépasser.
Abstract
Genetic markers are now widely used in the clinics, particularly in cancer patient management. Indeed, these tumor markers can help in the diagnosis and prognosis of the disease, and provide valuable information for treatment orientation in the context of personalized medicine. The presence of circulating cell-free tumor DNA (cftDNA) and thus of tumor markers in the blood can be considered to partly avoid the use of solid biopsies. The use of blood samples, as liquid biopsies, is less invasive and described as more representative of tumor heterogeneity. However, cftDNA can be found in blood in low proportion that can vary according to the nature and the progression of the tumor. For these reasons, the use of highly sensitive, specific and ideally quantitative methods for its detection are required. These requirements constituted until recently a technological limit, which now can be overcome thanks to digital PCR. This technology could now become a very efficient and non-invasive tool in oncology, complementary to conventional diagnostic techniques.
Le premier de ces deux articles a été publié dans m/s n° 1, vol 31, janvier 2015, page 84 [43].
© 2015 médecine/sciences – Inserm
Le développement et la démocratisation des techniques de biologie moléculaire ont permis d’importants progrès dans la détection et l’analyse des acides nucléiques. Ces techniques constituent aujourd’hui un outil clé de la prise en charge des patients atteints de cancer. En effet, des réseaux complexes d’altérations génétiques ont été mis en évidence dans la plupart des cancers. Il peut s’agir, entre autres, de délétions, de mutations ponctuelles, de réarrangements chromosomiques ou d’amplifications. Ces altérations génétiques étant présentes uniquement dans les cellules tumorales, elles représentent des marqueurs spécifiques de cancer. Ces marqueurs peuvent être classés en trois grandes catégories : il peut s’agir de marqueurs diagnostiques, permettant, par exemple, de déterminer la localisation de la maladie ou son stade ; de marqueurs pronostiques, permettant de détecter la progression ou la récurrence du cancer ; ou encore de marqueurs prédictifs, permettant de prédire la réponse aux traitements, la pharmacodynamique ou la toxicité de ces derniers [1]. Ces marqueurs, en caractérisant des tumeurs à l’échelle moléculaire, sont particulièrement pertinents dans le domaine de la médecine de précision. En effet, de nombreux traitements ciblés ont été développés ces dix dernières années, et de nouveaux marqueurs de sensibilité ou de résistance à ces traitements mis en évidence. Le suivi au cours du temps de ces altérations génétiques permettrait de suivre l’évolution du cancer, l’efficacité d’un traitement ou de détecter une éventuelle récidive. L’utilisation pleinement efficace de ces marqueurs en oncologie clinique nécessite cependant des méthodes précises et fiables pour les détecter dans différents types d’échantillons (biopsies tumorales, fluides biologiques dont le sang). L’avancée technologique récente que représente la PCR (polymerase chain reaction) digitale (dPCR) en microcompartiments permet la détection et la quantification de séquences rares avec une sensibilité et une précision hors de portée des procédures conventionnelles [43] (→). La dPCR a ainsi récemment émergé comme un outil extrêmement prometteur pour le suivi non invasif de marqueurs génétiques, particulièrement pertinent pour la recherche en cancérologie.
(→) Voir le Dossier technique de K. Perez-Toralla et al., m/s n° 1, janvier 2015, page 84
Caractéristiques de l’ADN libre circulant et contraintes liées à sa mesure
Des acides nucléiques sont retrouvés dans l’ensemble des fluides biologiques dont le sang, les urines, les selles, la salive ou le liquide synovial. La présence d’acides nucléiques dans le plasma a été montrée pour la première fois en 1948 [2], et de l’ADN libre circulant a été détecté en quantités importantes dès 1977 dans le sérum et le plasma de patients cancéreux [3]. L’ADN libre circulant est présent chez les sujets sains à des concentrations de l’ordre de 0 à 100 ng/ml [4]. Chez des patients atteints de cancer, ces concentrations varient de 0 à 5 000 ng/ml. Les ADN libres circulants se présentent sous forme de fragments d’ADN génomique, de tailles variables, libérés lors de l’apoptose, la nécrose, la lyse, ou la fragmentation de cellules [5, 6] (Figure 1). Les mesures de l’intégrité et de taux élevés d’ADN libre circulant dans le sang d’un patient ont été envisagées comme marqueurs de cancer [4], mais les résultats obtenus sont contradictoires. On observe, par exemple, une variabilité importante des quantités d’ADN libre circulant mesurées selon la durée écoulée depuis le prélèvement, l’état physiopathologique et le sexe du patient [5, 7].
 |
Figure 1. L’ADN libre circulant : de son origine à sa détection. A. Origine de l’ADN libre circulant. Les origines de l’ADN libre circulant sont multiples ; il peut s’agir d’ADN provenant de cellules tumorales, de cellules saines ou même de virus et de bactéries. L’ADN retrouvé dans le sang provient de cellules apoptotiques ou nécrotiques. B. Détection spécifique de l’ADN libre tumoral. L’analyse de l’ADN libre tumoral circulant nécessite des techniques de détection de plus en plus sensibles : PCR en temps réel dite quantitative (qPCR), séquençage de nouvelle génération (SNG), et PCR digitale (dPCR), cette dernière étant la plus sensible. |
Le temps de demi-vie moyen de l’ADN libre circulant a été estimé à 16 minutes dans le sang, en se basant sur des données de décroissance du taux d’ADN fœtal dans le plasma maternel [8]. Des travaux plus récents ont démontré que, chez des patients dont le plasma était prélevé de manière séquentielle après résection complète de leur tumeur, le temps de demi-vie de l’ADN libre tumoral circulant plasmatique (identifié grâce au suivi d’une altération spécifique de la tumeur) était de 114 min [9]. L’extraction d’ADN libre circulant doit donc idéalement être réalisée dans les heures suivant le prélèvement afin d’éviter toute dégradation. Le sang prélevé est centrifugé, puis la fraction plasmatique, ou la fraction sérique, sont concentrées. Le plasma est généralement préféré au sérum, car la collecte du sérum étant réalisée sans anticoagulant, un caillot se forme, source de dégradations cellulaires et de libération d’ADN (le taux d’ADN sérique est jusqu’à six fois supérieur aux taux d’ADN plasmatique). Les protocoles d’extraction de l’ADN libre circulant à partir du plasma peuvent différer d’un laboratoire à un autre, rendant difficile la comparaison des résultats. Cependant, des travaux ont été réalisés afin d’identifier les facteurs de variabilité de ces procédures [10].
Suivre directement l’ADN libre tumoral circulant est une possibilité très attractive pour la recherche en oncologie. La présence, dans le sang de patients atteints de cancer, d’ADN libre porteur d’altérations spécifiques a en effet été démontrée par de nombreux travaux [11]. Cet ADN tumoral, qui présente les mêmes signatures moléculaires que la tumeur, est libéré par les cellules tumorales lorsqu’elles meurent [4, 12]. Il a été proposé que le suivi cinétique de marqueurs génétiques circulants pourrait permettre de suivre l’évolution de la tumeur, de déterminer l’efficacité d’un traitement et de détecter d’éventuelles récidives (Figure 2) [13]. Ainsi, la chute des niveaux d’ADN libre tumoral circulant après résection complète de la tumeur et leur ré-augmentation lors du développement de nouvelles lésions ont été décrites [14]. Le suivi de marqueurs génétiques circulants pourrait également permettre d’éviter de multiples interventions chirurgicales ou l’analyse de prélèvements archivés de la tumeur primaire. De plus, les biopsies classiques, souvent difficiles, voire impossibles si la tumeur est inaccessible, peuvent faire courir un risque au patient, et être responsables de la dissémination de la tumeur [15].
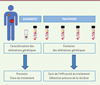 |
Figure 2. Suivi potentiel du statut des mutations à partir du sang. La recherche des altérations génétiques (mutations, amplifications ou translocations par exemple) caractérisant la tumeur (•) dans le sang pourrait aider au diagnostic, à l’orientation du traitement et à détecter les récidives éventuelles. Cette procédure est peu invasive, puisqu’elle ne nécessite qu’une simple prise de sang. La caractérisation moléculaire de la tumeur aide le clinicien au pronostic et au choix du traitement. Cependant, les altérations génétiques de la biopsie peuvent ne pas être représentatives de la tumeur elle-même (présence de cellules saines [•], de sous-clones minoritaires [•], etc.), ni d’éventuelles tumeurs secondaires ou métastases (•). L’ADN libre tumoral circulant (présent dans le sang) pourrait être plus représentatif de cette hétérogénéité et, par conséquent, sa signature moléculaire pourrait être plus pertinente, par exemple pour l’orientation du traitement. |
L’analyse du sang (biopsie « liquide ») pourrait également être plus représentative de l’état pathologique du patient [16]. En effet, l’ADN tumoral qui y circule pourrait refléter, à la fois l’hétérogénéité clonale de la tumeur, d’éventuelles métastases ou d’autres foyers tumoraux [11]. C’est un avantage sur les biopsies classiques qui, prélevées de manière ponctuelle et locale, peuvent sous-estimer cette diversité génétique [17].
L’ADN libre tumoral circulant portant des altérations spécifiques de la tumeur du patient peut cependant être très fortement dilué dans le pool d’ADN libre circulant provenant de la mort de cellules non porteuses de ces altérations. Il peut s’agir de cellules du microenvironnement tumoral comme de cellules saines de l’organisme [18]. Par exemple, l’ADN libre tumoral circulant peut représenter moins de 0,01 % de l’ADN libre circulant total pour des cancers à un stade précoce [14], et plus de 50 % pour des cancers métastatiques [13, 19, 20]. Ceci rend indispensable l’utilisation d’outils de détection plus sensibles et plus spécifiques que les techniques conventionnelles (PCR quantitative et séquençage). De plus, afin de réaliser un suivi efficace des taux d’ADN libre tumoral circulant, par exemple dans le cadre du suivi des patients atteints de cancer, les méthodes utilisées doivent également permettre une quantification précise de la quantité d’ADN libre circulant (ADN libre circulant total et tumoral) [21]. Les développements récents de la PCR digitale (dPCR) permettent aujourd’hui de répondre à ces exigences.
Contribution de la PCR digitale à la recherche en cancérologie
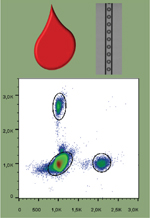
La dPCR permet la détection et la quantification absolue d’une séquence rare d’acides nucléiques diluée dans un large excès de séquences issues de cellules saines. La technique consiste à réaliser une réaction de PCR, non plus sur un mélange d’ADN cibles, mais sur des ADN cibles individuels, compartimentés par dilution limite de l’échantillon initial [43]. Chaque compartiment ne peut alors qu’être positif ou négatif pour la séquence ciblée1. Cela confère à cette procédure un caractère quantitatif, puisque chaque évènement (positif ou négatif) peut être compté. C’est de cette logique binaire que vient le terme de PCR « digitale ». La dPCR apporte ainsi un gain en sensibilité important par rapport aux techniques de PCR quantitative (qPCR) classiques (Figure 3). La sensibilité du test dépend du nombre de compartiments et de la quantité de molécules analysables, ainsi que du taux de faux-positifs inhérent à la technique utilisée [21]. Initialement réalisée en microplaques dès 1999 [22], la dPCR est désormais réalisée au sein de microcompartiments, dont le volume est de l’ordre du picolitre au nanolitre [21].
 |
Figure 3. Principe de la PCR digitale. La PCR digitale consiste à diluer un échantillon en un grand nombre de partitions et à compter le nombre de partitions dans lesquelles une réaction a lieu. Dans cet exemple, deux ADN sont ciblés avec des sondes portant des fluorophores différents (rouge ou vert). Modifié d’après Baker [42]. |
Les derniers développements de la dPCR en microcompartiments ont permis la détection de nombreux marqueurs génétiques dans le sang de patients atteints de cancer (Tableau I). La commercialisation, ces dernières années, d’appareils utilisant ces technologies, permet, entre autres, d’évaluer la pertinence de cette détection en recherche clinique, notamment celle de marqueurs tumoraux. La dPCR représente un outil innovant en oncologie clinique. Elle permet à la fois de réaliser plus efficacement les tests réalisés aujourd’hui par des méthodes telles que la qPCR, mais également de réaliser des expériences au-delà des capacités des procédures conventionnelles. Il est cependant à noter que de nouvelles procédures, plus sensibles que les procédures conventionnelles, basées sur la PCR quantitative spécifique d’allèles, ont été récemment développées pour l’analyse d’ADN libre tumoral circulant [23, 24] et validées dans le cadre d’études cliniques [19]. Ainsi, la dPCR est particulièremente pertinente pour la détection et la quantification de marqueurs minoritaires, incluant les sous-clones rares au sein de la tumeur ou de l’ADN libre circulant. Plusieurs études récentes ont ainsi montré son intérêt pour le suivi des cancers du côlon, du poumon et du sein à partir d’échantillons de sang [20, 25–27].
Exemples d’altérations génétiques tumorales ayant été détectées dans l’ADN libre circulant par PCR digitale. APC : anaphase promoting complex ; KRas : Kirsten rat sarcoma viral oncogene homolog ; PIK3CA : phosphoinositide-3-kinase, catalytic, a polypeptide ; TP53 : tumor protein 53 ; BRAF : v-raf murine sarcoma viral oncogene homolog B ; EGFR : epidermal growth factor receptor ; MET : met proto-oncogene ; IQCA1 : IQ motif containing with AAA domain 1 ; KIAA0406 : TELO2 interacting protein 1 ; ZFYVE21 : zinc finger, FYVE domain containing 21 ; ATM : ataxia telangiectasia mutated. Tableau non exhaustif, modifié et complété de Crowley et al. [11].
Trois exemples
Cancer colorectal : mutations de l’oncogène KRas
À titre d’exemple, dans le cas du cancer colorectal, des mutations de l’oncogène KRas (Kirsten rat sarcoma viral oncogene homolog) [44] sont associées à l’absence de réponse à des traitements ciblant le récepteur du facteur de croissance épidermique (EGFR) (anticorps monoclonaux cetuximab et panitumumab). Différents travaux se sont ainsi intéressés à la détection de mutations au sein de l’oncogène KRas chez des patients atteints de cancer colorectal. Dans une étude réalisée à l’aide de la technologie BEAMing2, l’émergence d’une résistance secondaire au cetuximab (KRAS p.Q61H) a pu être mise en évidence à partir de l’analyse de l’ADN libre circulant plasmatique, dix mois avant d’être détectée par des techniques d’imagerie médicale visualisant la tumeur [13]. Deux études, parues en 2011, présentent des technologies de dPCR utilisant des systèmes microfluidiques en microgouttelettes : l’une pour la détection de mutations de BRaf (v-raf murine sarcoma viral oncogene homolog B) avec une sensibilité de 0,001 % (soit un clone muté détecté parmi 100 000 clones non mutés) [28], et l’autre pour la détection de mutations de KRas avec une sensibilité similaire [29], suffisante pour la détection de mutations de KRas dans l’ADN libre circulant. Basée sur cette dernière technique, une procédure en multiplex permettant de détecter et de quantifier les sept mutations de KRas les plus répandues a été développée et utilisée pour caractériser l’ADN libre tumoral circulant plasmatique de patients atteints de cancer colorectal à un stade avancé (analyse faite lors de la progression tumorale et avant le début du traitement anti-EGFR). Dans cette étude (50 patients), la concordance entre le statut des mutations de l’ADN de la tumeur (déterminé par qPCR) et celui de l’ADN du plasma est de 86 % pour les expériences en multiplex, et de 90 % pour les expériences en duplex. Trois échantillons plasmatiques contenant un allèle KRas muté présentaient une discordance avec le profil de mutations de la tumeur du patient [20]. Des mutations de l’oncogène KRas indétectables dans les tissus tumoraux avaient également pu être mises en évidence à partir de sérum par dPCR BEAMing [30], illustrant la capacité des techniques de dPCR à refléter l’hétérogénéité clonale de la tumeur.
Cancer du poumon : mutations du gène codant pour l’EGFR
La prédiction d’éventuelles résistances peut aider le clinicien dans le choix du traitement le plus adapté, et le plus précoce possible. Par exemple, pour le cancer du poumon, deux mutations du gène codant pour l’EGFR (une délétion au sein de l’exon 19 ou la mutation ponctuelle L858R) confèrent une grande sensibilité des cellules tumorales à des molécules inhibitrices de l’EGFR (gefitinib et erlotinib, deux inhibiteurs sélectifs de la tyrosine kinase de EGFR), alors que la mutation T790M est identifiée comme une mutation secondaire de résistance à ces mêmes molécules. Les deux mutations associées à une sensibilité au traitement représentent 85 % des mutations de l’EGFR, et leur détection dans l’ADN libre tumoral circulant dans le plasma a pu être réalisée en dPCR (digital array chip 12.765®3) avec une sensibilité et une spécificité respectives de 92 % et 100 %. Chez quinze patients pour lesquels des mutations d’EGFR ont été détectées dans l’ADN libre tumoral circulant plasmatique, 40 % portaient la délétion et 60 % la mutation ponctuelle [31]. Ces proportions sont similaires à celles déterminées par des études réalisées sur le tissu tumoral, alors que, dans des études réalisées en qPCR à partir d’échantillons extraits de sérum, la mutation L858R est plus faiblement représentée [32]. La mutation T790M, habituellement identifiée chez environ la moitié des patients résistants traités avec les molécules mentionnées précédemment, a pu être détectée dans des échantillons plasmatiques par BEAMing chez 43,5 % des patients ayant une tumeur en progression après traitement [33].
Cancer du sein : amplification de HER2
Dans le cas du cancer du sein, les tumeurs d’entre 15 % et 25 % des patientes surexpriment HER2 (human epidermal growth factor receptor 2) par amplification du gène, et ces patientes doivent être traitées avec des thérapies ciblées (trastuzumab [anticorps herceptine] ou lapatinib [inhibiteur de l’acitvité tyrosine kinase]) pour améliorer leur pronostic. Ainsi, le suivi de cette amplification parmi les molécules d’ADN libre tumoral circulant permettrait de suivre au cours du temps l’efficacité du traitement. Dès 2011, l’amplification de ce gène a été détectée par qPCR dans des échantillons d’ADN libre circulant provenant du plasma de patients. Toutefois, chez 30 patientes atteintes de cancer du sein métastatique, la corrélation entre le taux d’amplification de HER2 détecté dans l’ADN libre circulant plasmatique et le statut d’HER2 de la tumeur primaire était faible [34]. Une étude parue plus récemment utilisant la dPCR, a été réalisée sur des échantillons de plasma provenant de 58 patientes présentant des métastases, et la concordance avec le statut de la tumeur primaire était de 90 % [27].
Conclusions
La multiplication des études réalisées sur l’ADN libre circulant utilisant, entre autres techniques, la dPCR, devrait contribuer à valider sa pertinence en tant que marqueur tumoral. Les résultats obtenus tendent à valider la dPCR en microcompartiments (microgouttelettes ou microchambres) pour le diagnostic, la détection de résistances et le suivi des patients pour, par exemple, anticiper une éventuelle récidive. Cette méthode offre une sensibilité et une spécificité supérieures à celles de la qPCR utilisée classiquement en oncologie clinique, ce qui permet la détection d’événements rares au sein de mélanges complexes d’ADN ayant de fortes similitudes de séquences avec la cible. Cela permet d’envisager le suivi de marqueurs tumoraux dans les liquides biologiques, tels que le sang, particulièrement adaptés au suivi des patients. La commercialisation récente d’appareils automatisant la dPCR va favoriser la démocratisation de ces analyses. Une grande partie des travaux réalisés initialement avaient pour objectif la détection de mutations spécifiques de tumeurs, mais de nouvelles stratégies pourraient permettre de détecter et quantifier d’autres types de marqueurs tumoraux. Des études en dPCR ont ainsi été décrites qui détectent les variations d’expression de différents microARN d’intérêt dans le sérum de patients atteints de cancer [35, 36]. De même, l’hyperméthylation du gène de la vimentine a pu être décelée par dPCR (BEAMing) dans l’ADN libre circulant plasmatique chez 59 % de patients atteints de cancer colorectal [37]. Les cellules tumorales circulantes sont également un marqueur d’intérêt [38] et peuvent être caractérisées par des procédures digitales. L’ADN y est déjà compartimenté, ce qui constitue un avantage par rapport à l’ADN libre circulant, mais leur manipulation est délicate et leur faible représentation impose des étapes de purification [39]. La possibilité de détecter des séquences génétiques spécifiques au sein de mélanges complexes, tels que les liquides biologiques, avec une haute sensibilité et spécificité, ouvre la voie à de nombreuses autres applications incluant, entre autres, le diagnostic viral [40] et le diagnostic prénatal [41].
Liens d’intérêt
V. Taly et P. Laurent-Puig déclarent participer à des travaux d’expertise pour la société RainDance.
Les autres auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Les auteurs remercient le Dr Deniz Pekin pour la relecture attentive de la revue. Ouriel Caen remercie l’ITMO Cancer de l’alliance nationale pour les sciences de la vie et de la santé (AVIESAN, Inserm, n°HAP_2012001) pour son financement accordé dans le cadre de l’école interdisciplinaire Frontières du vivant (FdV). Les auteurs remercient, pour leur soutien financier, le ministère de l’Enseignement supérieur et de la Recherche, l’université Paris-Descartes, l’université de Strasbourg, le Centre national de la recherche scientifique (CNRS), l’Institut national de la santé et de la recherche médicale (Inserm), l’Institut national du cancer (INCA, No. 2009-1-RT-03-US-1 and 2009-RT-03-UP5-1), l’Association pour la recherche contre le cancer (ARC, n° SL220100601375), l’Agence nationale de la recherche (ANR nanobiotechnologies), la région Alsace, et le SIRIC CARPEM.
Les procédures expérimentales peuvent toutefois imposer des compartimentations multiples. Dans ce cas, la distribution statistique de l’ADN dans les compartiments peut alors être évaluée par la loi de Poisson, qui régit les statistiques d’évènements rares.
Le BEAMing (beads, emulsion, amplification and magnetics) est une technologie commercialisée initialement par la société Ionostics (aujourd’hui par la société Sysmex). Elle utilise des billes magnétiques fonctionnalisées avec des amorces complémentaires de l’ADN cible qui, une fois « recouvertes » de multiples copies de l’ADN cible initialement encapsulé, sont marquées par des sondes fluorescentes spécifiques, puis analysées par cytométrie à fluorescence. Cette technique permet de réaliser des tests quantitatifs et très sensibles : une molécule d’ADN mutée peut être détectée parmi 10 000 molécules d’ADN
Le digital array chip 12.765® est une puce microfluidique commercialisée par la société Fluidigm. Elle permet d’analyser 12 échantillons différents en parallèle, chacun étant discrétisé en 765 compartiments fermés d’un volume de 6 nl. Cette technique permet de réaliser 9 180 réactions de PCR simultanées, avec une sensibilité d’une molécule d’ADN mutant détectée parmi mille molécules d’ADN non mutées.
Références
- Brooks JD. Translational genomics: the challenge of developing cancer biomarkers. Genome Res 2012 ; 22 : 183–187. [CrossRef] [PubMed] [Google Scholar]
- Mandel P, Metais P. Les acides nucléiques du plasma sanguin chez l’homme. CR Séances Soc Biol Fil 1948 ; 142 : 241–243. [Google Scholar]
- Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 1977 ; 37 : 646–650. [PubMed] [Google Scholar]
- Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 2011 ; 11 : 426–437. [CrossRef] [PubMed] [Google Scholar]
- Gormally E, Caboux E, Vineis P, Hainaut P. Circulating free DNA in plasma or serum as biomarker of carcinogenesis: practical aspects and biological significance. Mutat Res 2007 ; 635 : 105–117. [CrossRef] [PubMed] [Google Scholar]
- Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res 2001 ; 61 : 1659–1665. [PubMed] [Google Scholar]
- Van der Vaart M, Pretorius PJ. Is the role of circulating DNA as a biomarker of cancer being prematurely overrated? Clin Biochem 2010 ; 43 : 26–36. [CrossRef] [PubMed] [Google Scholar]
- Lo YMD, Zhang J, Leung TN, et al. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet 1999 ; 64 : 218–224. [CrossRef] [PubMed] [Google Scholar]
- Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008 ; 14 : 985–990. [CrossRef] [PubMed] [Google Scholar]
- El Messaoudi S, Rolet F, Mouliere F, Thierry AR. Circulating cell free DNA: preanalytical considerations. Clin Chim Acta 2013 ; 424 : 222–230. [CrossRef] [PubMed] [Google Scholar]
- Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol 2013 ; 10 : 472–484. [CrossRef] [Google Scholar]
- Stroun M, Lyautey J, Lederrey C, et al. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta 2001 ; 313 : 139–142. [CrossRef] [PubMed] [Google Scholar]
- Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012 ; 486 : 532–536. [CrossRef] [PubMed] [Google Scholar]
- Lecomte T, Berger A, Zinzindohoue F, et al. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int J Cancer 2002 ; 100 : 542–548. [CrossRef] [PubMed] [Google Scholar]
- Robertson EG, Baxter G. Tumour seeding following percutaneous needle biopsy: the real story!. Clin Radiol 2011 ; 66 : 1007–1014. [CrossRef] [PubMed] [Google Scholar]
- Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature 2013 ; 501 : 355–364. [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012 ; 366 : 883–892. [CrossRef] [PubMed] [Google Scholar]
- Diehl F, Li M, Dressman D, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA 2005 ; 102 : 16368–16373. [Google Scholar]
- Thierry AR, Mouliere F, El Messaoudi S, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med 2014 ; 20 : 430–435. [CrossRef] [PubMed] [Google Scholar]
- Taly V, Pekin D, Benhaim L, et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem 2013 ; 59 : 1722–1731. [CrossRef] [PubMed] [Google Scholar]
- Taly V, Pekin D, El Abed A, Laurent-Puig P. Detecting biomarkers with microdroplet technology. Trends Mol Med 2012 ; 18 : 405–416. [CrossRef] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci USA 1999 ; 96 : 9236–9241. [CrossRef] [Google Scholar]
- Thierry AR, Mouliere F, Gongora C, et al. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res 2010 ; 38 : 6159–6175. [CrossRef] [PubMed] [Google Scholar]
- Didelot A, Kotsopoulos SK, Lupo A, et al. Multiplex picoliter-droplet digital PCR for quantitative assessment of DNA integrity in clinical samples. Clin Chem 2013 ; 59 : 815–823. [CrossRef] [PubMed] [Google Scholar]
- Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res 2014 ; 20 : 1698–1705. [CrossRef] [PubMed] [Google Scholar]
- Dawson SJ, Tsui DWY, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 2013 ; 368 : 1199–1209. [CrossRef] [PubMed] [Google Scholar]
- Gevensleben H, Garcia-Murillas I, Graeser MK, et al. Noninvasive detection of HER2 amplification with plasma DNA digital PCR. Clin Cancer Res 2013 ; 19 : 3276–3284. [CrossRef] [PubMed] [Google Scholar]
- Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 2011 ; 83 : 8604–8610. [CrossRef] [PubMed] [Google Scholar]
- Pekin D, Skhiri Y, Baret JC, et al. Quantitative and sensitive detection of rare mutations using droplet-based microfluidics. Lab Chip 2011 ; 11 : 2156–2166. [CrossRef] [PubMed] [Google Scholar]
- Diaz LA Jr,Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012 ; 486 : 537–540. [CrossRef] [PubMed] [Google Scholar]
- Yung TKF, Chan KCA, Mok TSK, et al. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin Cancer Res 2009 ; 15 : 2076–2084. [CrossRef] [PubMed] [Google Scholar]
- Kimura H, Kasahara K, Shibata K, et al. EGFR mutation of tumor and serum in gefitinib-treated patients with chemotherapy-naive non-small cell lung cancer. J Thorac Oncol 2006 ; 1 : 260–267. [Google Scholar]
- Taniguchi K, Uchida J, Nishino K, et al. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res 2011 ; 17 : 7808–7815. [CrossRef] [PubMed] [Google Scholar]
- Page K, Hava N, Ward B, et al. Detection of HER2 amplification in circulating free DNA in patients with breast cancer. Br J Cancer 2011 ; 104 : 1342–1348. [CrossRef] [PubMed] [Google Scholar]
- Moltzahn F, Olshen AB, Baehner L, et al. Microfluidic-based multiplex qRT-PCR identifies diagnostic and prognostic microRNA signatures in the sera of prostate cancer patients. Cancer Res 2011 ; 71 : 550–560. [CrossRef] [PubMed] [Google Scholar]
- Ma J, Li N, Guarnera M, Jiang F. Quantification of plasma miRNAs by digital PCR for cancer diagnosis. Biomark Insights 2013 ; 8 : 127–136. [PubMed] [Google Scholar]
- Li M, Chen WD, Papadopoulos N, et al. Sensitive digital quantification of DNA methylation in clinical samples. Nat Biotechnol 2009 ; 27 : 858–863. [CrossRef] [PubMed] [Google Scholar]
- Riethdorf S, Fritsche H, Muller V, et al. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the cell search system. Clin Cancer Res 2007 ; 13 : 920–928. [CrossRef] [PubMed] [Google Scholar]
- Pfitzner C, Schröder I, Scheungraber C, et al. Digital-direct-RT-PCR: a sensitive and specific method for quantification of CTC in patients with cervical carcinoma. Sci Rep 2014 ; 4 : 3970. [CrossRef] [PubMed] [Google Scholar]
- Sedlak RH, Jerome KR. Viral diagnostics in the era of digital polymerase chain reaction. Diagn Microbiol Infect Dis 2012 ; 75 : 1–4. [CrossRef] [PubMed] [Google Scholar]
- Lo YM, Chiu RW. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidies by maternal plasma nucleic acid analysis. Clin Chem 2008 ; 54 : 461–466. [CrossRef] [PubMed] [Google Scholar]
- Baker M.. Digital PCR hits its stride. Nat Meth 2012 ; 9 : 541–544. [CrossRef] [Google Scholar]
- Perez-Toralla K, Pekin D, Bartolo JF, et al. PCR digitale en microcompartiments : détection sensible de séquences d’acides nucléiques rares. Med sci (Paris) 2015 : 84–92. [Google Scholar]
- Bournet B, Dufresne M, Selves J, et al. Oncogène Kras et cancer du pancréas. Med Sci (Paris) 2013 ; 29 : 991–997. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des tableaux
Exemples d’altérations génétiques tumorales ayant été détectées dans l’ADN libre circulant par PCR digitale. APC : anaphase promoting complex ; KRas : Kirsten rat sarcoma viral oncogene homolog ; PIK3CA : phosphoinositide-3-kinase, catalytic, a polypeptide ; TP53 : tumor protein 53 ; BRAF : v-raf murine sarcoma viral oncogene homolog B ; EGFR : epidermal growth factor receptor ; MET : met proto-oncogene ; IQCA1 : IQ motif containing with AAA domain 1 ; KIAA0406 : TELO2 interacting protein 1 ; ZFYVE21 : zinc finger, FYVE domain containing 21 ; ATM : ataxia telangiectasia mutated. Tableau non exhaustif, modifié et complété de Crowley et al. [11].
Liste des figures
|
Figure 1. L’ADN libre circulant : de son origine à sa détection. A. Origine de l’ADN libre circulant. Les origines de l’ADN libre circulant sont multiples ; il peut s’agir d’ADN provenant de cellules tumorales, de cellules saines ou même de virus et de bactéries. L’ADN retrouvé dans le sang provient de cellules apoptotiques ou nécrotiques. B. Détection spécifique de l’ADN libre tumoral. L’analyse de l’ADN libre tumoral circulant nécessite des techniques de détection de plus en plus sensibles : PCR en temps réel dite quantitative (qPCR), séquençage de nouvelle génération (SNG), et PCR digitale (dPCR), cette dernière étant la plus sensible. |
| Dans le texte | |
|
Figure 2. Suivi potentiel du statut des mutations à partir du sang. La recherche des altérations génétiques (mutations, amplifications ou translocations par exemple) caractérisant la tumeur (•) dans le sang pourrait aider au diagnostic, à l’orientation du traitement et à détecter les récidives éventuelles. Cette procédure est peu invasive, puisqu’elle ne nécessite qu’une simple prise de sang. La caractérisation moléculaire de la tumeur aide le clinicien au pronostic et au choix du traitement. Cependant, les altérations génétiques de la biopsie peuvent ne pas être représentatives de la tumeur elle-même (présence de cellules saines [•], de sous-clones minoritaires [•], etc.), ni d’éventuelles tumeurs secondaires ou métastases (•). L’ADN libre tumoral circulant (présent dans le sang) pourrait être plus représentatif de cette hétérogénéité et, par conséquent, sa signature moléculaire pourrait être plus pertinente, par exemple pour l’orientation du traitement. |
| Dans le texte | |
|
Figure 3. Principe de la PCR digitale. La PCR digitale consiste à diluer un échantillon en un grand nombre de partitions et à compter le nombre de partitions dans lesquelles une réaction a lieu. Dans cet exemple, deux ADN sont ciblés avec des sondes portant des fluorophores différents (rouge ou vert). Modifié d’après Baker [42]. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.