")
")
| Issue |
Med Sci (Paris)
Volume 31, Number 2, Février 2015
|
|
|---|---|---|
| Page(s) | 143 - 150 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20153102010 | |
| Published online | 4 mars 2015 | |
Dérégulation de l’hémostase dans les infections à filovirus
Haemostasis dysregulation in filovirus infections
CIRI (centre international de recherche en infectiologie), Inserm U1111, laboratoire bases moléculaires de la pathogénicité virale, 21, avenue Tony Garnier, 69007 Lyon, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les filovirus sont responsables d’infections à fort taux de létalité. Ces virus sont retrouvés dans les régions intertropicales d’Afrique et d’Asie ou ils circulent dans leurs réservoirs présumés, les chauves-souris frugivores. Lors des épidémies à filovirus, des modifications de l’hémostase ont pu être rapportées chez les patients, exprimées à des degrés différents selon les souches. Parmi celles-ci, la coagulation intravasculaire disséminée est une caractéristique majeure : elle est multifactorielle et met en jeu des facteurs viraux et une réponse physiologique inadéquate. Dans cette revue, nous décrirons les mécanismes mis en jeu dans ces dérèglements et discuterons les bases de la pathogénicité de ces virus.
Abstract
Filoviruses are responsible for highly lethal infections. Those viruses are found in intertropical areas of Africa and Asia where they circulate in their supposed natural reservoir, fruit bats. During filovirus outbreaks and depending on the strains, various modifications in hemostasis have been observed in patients. The disseminated intravascular coagulation identified in these infections is multicausal and involves both viral factors and abnormal physiological responses. In this review we will describe the mechanisms responsible for these disturbances and we will highlight some aspects of the basis of filovirus high pathogenicity.
Vignette (Photo © Inserm-Peter Lenting).
© 2015 médecine/sciences – Inserm
La famille des Filoviridae et leurs réservoirs
Depuis le mois de mars 2014, la plus grosse épidémie due au virus Ebola touche l’Afrique de l’Ouest au travers de la Guinée, du Libéria et de la Sierra Leone. Ce virus, tout comme le virus Marburg, appartient à la famille des Filoviridae. Ils doivent leur nom à la morphologie filiforme des virions. Ce sont des agents pathogènes de niveau 4, responsables de fièvres hémorragiques virales (FHV) souvent fatales chez les primates humains et non humains. Il n’existe à l’heure actuelle aucun traitement curatif ou prophylactique commercialement disponible capable d’endiguer ces infections. Dans cette revue, nous nous attacherons à décrire les multiples mécanismes des dérégulations physiologiques observées dans les infections à filovirus.
Ces filovirus appartiennent à l’ordre des Mononegavirales, qui regroupe les virus à ARN négatif simple brin non segmentés. Les genres Marburgvirus, Ebolavirus et Cuevavirus constituent la famille des Filoviridae. Il existe cinq espèces de virus Ebola : Zaïre, Reston, Bundibugyo, Sudan et Taï Forest ; deux espèces de virus Marburg : les virus Marburg et Ravn ; et une seule espèce décrite de Cuevavirus, le virus Lloviu [1].
Les filovirus ont en commun la structure de leur génome. Celle-ci est composée d’une succession linéaire de gènes, séparée par de courtes régions intergéniques et codant pour la nucléoprotéine NP, la protéine VP35, la protéine VP40, la glycoprotéine sécrétée sGP et la glycoprotéine de surface (GP), la protéine VP30, la protéine VP24 et la polymérase L [2, 3]. Les protéines NP, VP35, VP30 et L constituent, avec l’ARN viral, le complexe réplicatif permettant la synthèse des ARN messagers viraux et la production d’ARN génomique et anti-génomique viral. Ce complexe, appelé ribonucléoprotéine, est transporté à la membrane plasmique. L’interaction avec la protéine de matrice VP40 déclenche le bourgeonnement des virions à la surface cellulaire. Ceux-ci sont ainsi enveloppés d’une bicouche lipidique et arborent à leur surface la glycoprotéine GP, responsable de l’attachement aux cellules cibles. Cette GP provient de l’édition du gène codant pour la sGP, un phénomène exclusivement retrouvé dans les virus Ebola et absent des virus Marburg [3]. Fait intéressant, bien que les protéines des virus Ebola et Marburg soient très similaires, leurs fonctions diffèrent. Ainsi, chez Ebola, la protéine VP24 bloque la réponse à l’interféron, alors que cette fonction est portée par la protéine de matrice VP40 dans le virus Marburg [4, 5]. La protéine VP24 du virus Marburg a, quant à elle, été récemment identifiée comme un effecteur important dans la régulation du stress oxydatif, par sa liaison à Keap1 (kelch-like ECH-associated protein 1) [6].
Depuis la découverte du premier filovirus (Marburg 1967), de nombreuses campagnes de recherche ont été entreprises afin de caractériser le réservoir de ces virus. Ces études ont permis d’identifier divers mammifères infectés par le virus Ebola et notamment des chauves-souris [7, 8]. En 2005, Éric Leroy et son équipe ont identifié l’ARN viral dans des organes de chauve-souris frugivores ne présentant aucun symptôme visible [9]. Trois espèces sont supposées être les réservoirs du virus Ebola : Hypsignathus monstrosus, Epomops franqueti et Myonycteris torquata (Figure 1). Cependant, il n’a jamais été possible de confirmer formellement la présence de virus infectieux chez ces espèces. À l’inverse, la circulation du virus Marburg dans les populations de Rousettus aegyptiacus et un lien direct entre les souches isolées de ces animaux et celle responsable d’épidémies chez l’homme ont pu être établis [10]. Les animaux porteurs de ce virus ne présentaient pas de symptôme apparent, montrant ainsi que cette espèce contrôle parfaitement la réplication du virus. Quant au virus Lloviu, son ARN a été découvert très récemment en Espagne lors d’une épidémie ayant décimé une population de chiroptères, les minioptères de Schreiber, en 2002 [11]. Aucun élément ne permet cependant d’affirmer que ce virus est responsable de la mortalité observée chez ces chauves-souris. Cette observation est la première réalisée chez des chauves-souris insectivores, ce qui sous-entend que les filovirus pourraient être retrouvés à plus grande échelle et dans des espèces beaucoup plus diverses.
 |
Figure 1. Les chauves-souris, réservoirs présumés des filovirus. A. Hypsignathus monstrosus. B. Epomops franqueti. C. Myonycteris torquata. D. Rousettus aegyptiacus. E. Miniopterus Schreibersi. |
Manifestations cliniques de l’infection à filovirus
Chez l’homme, les symptômes cliniques apparaissent après une phase d’incubation de 2 à 21 jours ; ils sont non spécifiques au début, sous la forme d’une brusque poussée de fièvre et de forts maux de tête qui durent 5 à 7 jours (Figure 2). Les patients souffrent le plus souvent de cachexie, de vomissements, de diarrhée, de douleurs abdominales et d’une altération de leur état mental. Dans 10 à 50 % des cas, des symptômes hémorragiques apparaissent sous forme d’hémorragies pétéchiales, de saignements au niveau du tractus digestif, des conjonctives, et très souvent aux points d’injections [12, 13]. Selon les épidémies et le type de virus, la présence de symptômes hémorragiques est variable. Notamment, il semble que dans l’épidémie actuelle en Afrique de l’Ouest, ceux-ci soient moins fréquents qu’à l’accoutumé [6]. Dans les infections fatales, le décès est lié de façon générale à une incapacité de la réponse immunitaire à contrôler la croissance virale, ce qui induit une défaillance multiviscérale, une dérégulation du système cardiovasculaire (pression sanguine, biodistribution des fluides) et une dérégulation de l’hémostase [15]. En dépit de l’extrême pathogénicité du virus Ebola, des patients infectés survivent à la phase aiguë de la maladie et guérissent de l’infection en absence de tout traitement, en contrôlant la réplication virale. Une réponse inflammatoire précoce et mesurée semble avoir un rôle dans le contrôle de l’infection par le virus Ebola [16]. Cependant, les mécanismes moléculaires mis en jeu sont peu connus. De plus, lors des épidémies de virus Ebola, des cas d’infections humaines asymptomatiques ont été identifiés chez des individus directement exposés à des patients ou à du matériel contaminé par le virus Ebola [17, 18]. Cette observation est à rapprocher du fait que la plupart des mammifères non primates infectés expérimentalement développent également une infection asymptomatique [8]. Ainsi, il existe probablement des facteurs génétiques ou immunitaires qui permettent de résister à l’infection.
 |
Figure 2. Description de la fièvre hémorragique à virus Ebola. Schéma du développement des fievres hémorragiques à virus Ebola à partir des données des épidemies de Mosango, de Yambuku et Gulu (données OMS et [21]). |
La contamination par le virus se fait généralement au niveau des muqueuses (digestives, oculaires, etc.) ou via des lésions de la peau. Des études détaillées des infections à filovirus ont montré que les cellules dendritiques et les macrophages sont, dans ces tissus, les cellules cibles initiales [19–21]. Ces cellules mobiles répliquent activement le virus et transportent l’agent pathogène vers les ganglions lymphatiques régionaux, d’où, après d’autres cycles réplicatifs, des virions sont libérés par les voies lymphatiques et la circulation sanguine. Par la suite, les organes principalement infectés sont la rate et le foie. Dans ces tissus, le virus infecte les cellules parenchymateuses, y compris les hépatocytes et les cellules corticales surrénales, entraînant l’expansion des foyers de nécrose dus à la mort cellulaire viro-induite. Les conclusions des quelques rapports disponibles d’autopsie de patients infectés font état d’une hémorragie limitée dans les muqueuses, la peau et les organes parenchymateux, y compris l’estomac et les intestins, associée à de vastes zones de nécrose du parenchyme hépatique [22–25]. Des nécroses similaires, avec dépôt de fibrine, ont également été décrites dans la pulpe blanche et la pulpe rouge de la rate, modifiant souvent l’architecture de cet organe. De plus, bien que les cellules endothéliales en culture soient facilement infectées par le virus, il y a peu de preuves d’une réplication virale dans ces cellules in vivo avant les dernières étapes de la maladie [26].
Une nécrose multifocale est observée dans les tissus lymphoïdes ; celle-ci ne résulte pas directement de l’infection par le virus - les lymphocytes ne sont pas infectables -, mais provient d’un emballement du système immunitaire, qui, lui, est responsable de l’apoptose massive des lymphocytes [16, 17, 21, 27].
Durant l’évolution de la maladie, on note des modifications hématologiques - lymphopénie, hyperleucocytose - et biochimiques, dont une élévation des concentrations sériques d’aspartate et d’alanine aminotransférases, caractéristique de l’atteinte hépatique [28]. Une thrombocytopénie, associée à une augmentation des D-dimères (produits de dégradation de la fibrine), et une élévation du temps de céphaline activée sont retrouvées durant la phase aiguë de l’infection. Ces modifications reflètent la coagulation intravasculaire disséminée (CIVD) associée au syndrome hémorragique. Cette CIVD est une caractéristique majeure de l’infection à filovirus.
La CIVD, une origine multifactorielle
Ce syndrome se caractérise par la formation localisée de multiples caillots dans la circulation sanguine, ce qui a pour effet de « consommer » les facteurs de coagulation et les plaquettes. Ces thromboses localisées entraînent des défaillances fonctionnelles, et, simultanément, un déficit de la coagulation sanguine par « consommation » des facteurs de coagulation. Une CIVD est retrouvée chez l’homme atteint de fièvres hémorragiques à filovirus, ainsi que chez les primates non-humains infectés, mais pas chez les rongeurs, à l’exception du hamster [29]. Si la survenue d’une CIVD semble donc dépendre de l’espèce, la dérégulation du système immunitaire, elle, caractérise tous les modèles animaux et joue un rôle majeur dans l’absence de contrôle de la réplication virale. L’origine de la CIVD est multifactorielle ; certains facteurs sont liés directement aux virus, d’autres proviennent des dommages induits et de l’emballement de la réponse inflammatoire.
Le facteur tissulaire
Les études faites dans un modèle primate infecté par le virus Ebola Zaïre ont montré que les anomalies de la coagulation surviennent en tout début d’infection, lorsque les macrophages infectés expriment le facteur tissulaire (FT) à leur surface [30]. Cette glycoprotéine de 47 kDa est un récepteur transmembranaire exprimé par les épithéliums, les muqueuses, la capsule des organes et, de manière inductible, par les macrophages, les monocytes et les cellules endothéliales. Le facteur tissulaire est exprimé dans des compartiments qui ne sont pas normalement en contact avec le sang. Il joue un rôle de senseur des hémorragies, et induit la cascade de coagulation du sang lors de lésions qui mettent en contact le sang et les tissus. Cette protéine est également retrouvée, de façon pathologique, dans la circulation sanguine, à la surface de microparticules membranaires, et ce notamment en cas de CIVD. Ces microparticules sont notamment libérées par les monocytes. La stimulation des monocytes par des endotoxines, du TNF (tumor necrosis factor), de l’IL(interleukine)-1β ou encore par MCP-1 (monocyte chimoattractant protein 1), induit l’expression à leur surface du facteur tissulaire [31, 32]. La libération de facteur tissulaire actif dans la circulation contribue directement à la formation de thrombus [33]. En effet, le facteur tissulaire s’associe au facteur VIIa pour former un complexe protéolytique ; ces complexes FT- FVIIa activent d’autres molécules de FVII, ainsi que les FIX (le FIXa catalyse la formation de FXa) et FX, essentiels à la protéolyse massive de prothrombine en thrombine (Voir la figure 2 de [56]) (Figure 3). L’action de la thrombine sur le fibrinogène induit la génération de dépôts de fibrine, la formation de multiples thrombus et la consommation des facteurs de coagulation. Cette activation de la voie du facteur tissulaire - dite intrinsèque - conduit également à la consommation des plaquettes. De plus, le complexe FT-FVIIa est un élément de signalisation connu notamment pour induire dans les cellules un afflux de Ca2+ ; c’est aussi un facteur pro-inflammatoire (via l’induction d’IL-6 et d’IL-8) [34].
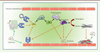 |
Figure 3. Rôle du facteur tissulaire dans l’initiation de la coagulation. L’association du facteur tissulaire (FT) avec le facteur VIIa constitue l’élément initiateur de nombreuses voies d’activation et notamment celle de l’activation du fibrinogène. |
Différentes protéines recombinantes (rNAPC2 et APC/Xigris) inhibitrices de la voie du facteur tissulaire ont montré leur efficacité dans le traitement de l’infection à virus Ebola chez le singe [35, 36]. Le rNAPC2 (pour recombinant nematode anticoagulant protein c2) inhibe directement l’activité du complexe FT-FVIIA en se fixant sur le facteur X. Le Xigris, lui, est une forme recombinante de la protéine C activée1 (APC). Cette protéine recombinante est indiquée dans le traitement des chocs septiques quelle qu’en soit la cause [57]. La protéine C activée est un élément clé de la régulation de la coagulation et, dans des modèles primates infectés, une chute de son taux a été observée [30, 35]. Cependant, une récente étude a montré que pendant l’épidémie à virus Ebola Sudan de 2000-2001, le taux de facteur tissulaire n’était pas élevé, et il n’y avait pas de corrélation entre ce taux et la présence de signes hémorragiques ou la létalité [37]. Les différences observées entre le modèle primate et l’homme pourraient être liées à des facteurs spécifiques de l’hôte ou du virus (virus Ebola Zaïre dans l’étude primate).
Atteinte hépatique
Les filovirus sont des virus majoritairement hépatotropes. Les rares études histologiques d’échantillons hépatiques provenant de patients humains infectés par le virus Zaïre ont montré une nécrose focale impliquant une ou plusieurs cellules [38]. La séquence qui aboutit à la nécrose des hépatocytes se caractérise d’abord par une éosinophilie cytoplasmique avec un obscurcissement, voire la dissolution, des noyaux, puis finalement une raréfaction des noyaux associée à une cytolyse, laissant la place à de grandes quantités de débris karyorrhectiques. Fait intéressant, compte tenu de l’ampleur des zones de nécroses observées dans cette étude, l’infiltration inflammatoire dans les sinusoïdes est très réduite. L’analyse des échantillons provenant de patients infectés par le virus Marburg montre des lésions très similaires, mais la nécrose hépatique ne semble pas suivre la séquence décrite lors de l’infection par Ebola [38]. Ainsi, l’infection massive et la nécrose du parenchyme hépatique induisent une modification de la fonction hépatique et affectent notamment la production des facteurs de coagulation, empêchant très certainement la restauration de taux physiologiques de facteurs de coagulation consommés lors de la CIVD, et aggravant le trouble de l’hémostase.
Libération anarchique de cytokines
Une des caractéristiques des infections à filovirus est la libération désordonnée de cytokines pro- et anti-inflammatoires, et notamment d’IFN (interféron) de type I, de TNFα, d’IL-1, IL-2, IL-6, IL-8, IL-10 et d’IL1RA (interleukin-1 receptor antagonist). Les données bibliographiques concernant le profil des cytokines sont toutefois encore incomplètes et parfois contradictoires (IFN et TNFα), et semblent varier selon l’espèce de virus Ebola [27, 37, 39, 40]. De plus, les données des concentrations de cytokines sériques représentent la somme des sécrétions par différents types cellulaires et dans différents compartiments ; ceci peut expliquer en partie les différences entre les observations in vitro et in vivo. Le TNFα est sécrété en abondance par les macrophages infectés in vitro [41], et sa sécrétion est inhibée dans les cellules dendritiques dérivées de monocytes [42]. Il est intéressant de noter que plusieurs publications montrent in vitro le rôle de la glycoprotéine de surface virale (sous forme de VLP) dans l’induction de la sécrétion de TNFα par les cellules dendritiques [43, 44]. Ce dernier est un puissant stimulant des cellules endothéliales, engendrant l’expression du facteur tissulaire à leur surface, qui peut déclencher la cascade de coagulation. De plus, l’IL-6 et l’IL-1 sont aussi actives dans l’induction de la coagulation [45, 46]. Une telle libération de cytokines devrait également libérer le facteur d’agrégation plaquettaire2 (ou PAF) et des éicosanoïdes (dont le thromboxane, puissant inducteur de l’agrégation plaquettaire). La libération massive de cytokines inflammatoires et l’atteinte hépatique engendrent à leur tour la libération de quantités importantes de monoxyde d’azote (NO). En effet, lors de l’épidémie à virus Ebola Sudan en 2000, des taux de plus de 150 µm de NO ont pu être mesurés 8 jours après le début des symptômes [47]. Le monoxyde d’azote a pour principal effet le relâchement de la tunique de cellules musculaires lisses des vaisseaux sanguins. Une libération importante comme celle qui est observée lors de la phase aiguë de l’infection entraîne une vasodilatation massive associée à une chute de la pression artérielle et une détresse cardiaque (des symptômes observés en phase aiguë de l’infection). Cependant, le monoxyde d’azote exerce aussi un rôle protecteur sur l’endothélium, et sa présence en quantité importante peut aussi contribuer à préserver l’intégrité de l’endothélium ; cela pourrait expliquer l’absence d’altération vasculaire dans les infections à filovirus [48].
Cellules endothéliales
Les cellules endothéliales constituent la barrière entre le sang et les tissus, elles assurent l’échange contrôlé des molécules biologiques via un ensemble de jonctions intercellulaires jouant un rôle de barrière pour l’eau et les sels (voie paracellulaire), et via une voie transcellulaire pour les macromolécules. Les données concernant le rôle des cellules endothéliales dans les infections à filovirus sont contradictoires. En effet, ces cellules peuvent répliquer le virus et s’activer, cependant leur infection in vivo n’a été montrée chez le singe qu’à des stades très tardifs de l’infection [20]. De plus, il n’a pas été noté post-mortem chez le singe et l’homme de dommages vasculaires notables. L’activation de cette barrière naturelle par le TNFα produit par les cellules infectées devrait conduire à un remodelage de leur architecture, entraînant une fuite de liquide vers l’interstitium. Étonnamment, la sGP, une protéine spécifique au virus Ebola, semble exercer un rôle protecteur vis-à-vis des effets du TNFα sur ces cellules. La glycoprotéine structurale a, quant à elle, montré des propriétés inverses [26]. De plus, l’activation des cellules endothéliales induit l’expression à leur surface du facteur tissulaire, qui, comme nous l’avons décrit précédemment, déclenche la coagulation. Ainsi, ces cellules pourraient participer à la mise en place d’une CIVD, sans toutefois subir de modification majeure de leur architecture. Cependant, la plupart des données disponibles sur le comportement des cellules endothéliales lors de l’infection par Ebola relèvent de modèles in vitro, et des études complémentaires in vivo seraient nécessaires afin de mieux identifier leur rôle dans les fièvres hémorragiques à filovirus.
Charges virales
Lors de la phase aiguë d’une infection à virus Ebola, les charges virales sanguines chez les patients atteignent 106,5 pfu/ml. Ces particules arborent à leur surface la glycoprotéine d’enveloppe (GP). Cette protéine fortement glycosylée a un pouvoir activateur sur les cellules dendritiques et les macrophages, grâce à son domaine mucinique [49]. Dans le modèle du cobaye, la forme soluble (shedGP) et la forme alternative (sGP) de la glycoprotéine virale sont présentes en très fortes quantités dans le sérum des animaux infectés. Une étude récente a montré que la forme libre de la glycoprotéine de surface (shedGP) était capable d’induire la libération massive de cytokines par les cellules dendritiques et les macrophages, via la stimulation du récepteur TLR4 (toll like receptor), mais également d’induire in vitro une altération des cellules endothéliales provoquant leur perméabilisation [50]. Enfin, les glycosylations de cette protéine constituent un excellent ligand pour les lectines sériques (notamment la MBL [mannose-binding lectin]) [51]. Cette lectine est un des acteurs majeurs de la voie d’activation du complément et de l’activation de la coagulation, via le recrutement des sérine protéases sériques (MASP [mannose-binding protein-associated serine protease]) [52, 53].
Conclusion
Comme nous l’avons montré, les causes du dérèglement de l’hémostase observé dans les infections à filovirus sont multiples et intriquées. Bien que les symptômes hémorragiques n’entraînent en général pas de perte volumique suffisante pour être la cause directe du décès, ils sont cependant la signature caractéristique de ce type d’infection. Le dérèglement systématique de l’hémostase en phase aiguë de l’infection est un élément clé de l’instauration du choc septique observé pendant les fièvres hémoragiques virales (Figure 4).
 |
Figure 4. Origine multifactorielle du dérèglement de l’hémostase dans l’infection à filovirus. L’infection virale conduit au dérèglement de nombreux mécanismes qui par synergie résulte en l’instauration d’une CIVD et d’un état de choc. |
La récente étude de McElroy et collaborateurs [37] a mis en évidence pour l’épidémie à virus Ebola Sudan que la présence de symptômes hémorragiques était liée statistiquement à une hausse de la ferritine, de la thrombomoduline et du facteur soluble d’adhésion intracellulaire (sICAM), mais qu’il n’existait pas de corrélation avec le taux de facteur tissulaire. La connaissance des mécanismes impliqués dans l’activation du facteur tissulaire par certaines enzymes extracellulaires (par exemple PDI, protéine disulfure isomérase) reste encore peu explorée dans le cas des fièvres hémorragiques et constitue une thématique de recherche prometteuse. L’étude des interactions entre la glycoprotéine du virus Ebola et les lectines du sérum pourrait également aider à une meilleure compréhension de la pathologie et servir de marqueur prédictif. Ainsi, il existe par exemple une très grande variabilité interindividuelle du taux sérique de la MBL, liée à des différences de génotype.
Les différences observées entre les données in vitro, in vivo et épidémiologiques montrent à quel point il est important de clarifier les mécanismes exacts de la pathogenèse des filovirus. L’épidémie en cours en Afrique de l’Ouest révèle également certaines particularités : la symptomatologie est légèrement différente de celle qui a été observée dans le passé, et notamment la fréquence de symptômes hémorragiques est plus faible [14]. Dans le futur, il serait intéressant d’étudier les bases moléculaires de cette différence de pathogénicité.
Pour conclure brièvement sur le traitement des infections à filovirus, il semble qu’il doive se baser sur trois approches parallèles ; un traitement de support visant à rétablir les constantes vitales (hydratation, pression artérielle, paramètres hématologiques), un traitement antiviral (ARNi [TKM-Ebola], des inhibiteurs du cycle viral (Favipiravir), des anticorps monoclonaux [ZMApp]) et, enfin, un traitement visant à réguler les mécanismes physiologiques (rNapC2 et Xigris évoqués précédemment, héparine, plasmaphérèse, etc.). Cette dernière approche a montré des résultats encourageants dans des modèles primates (diminution de 30 % de la mortalité avec le rNapC2). De plus, les traitements de support qui font actuellement l’objet d’études par de nombreuses équipes médicales dans les cas des fièvres hémorragiques dues à la Dengue, pourraient s’avérer potentiellement transposables aux infections à filovirus [54, 55].
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
« La protéine C est une protéine dont la synthèse dépend de la vitamine K. Physiologiquement, elle circule très majoritairement sous la forme d’un zymogène inactif et acquiert une activité sérine protéase une fois clivée par la thrombine. C’est donc la thrombine formée lors de l’activation de la coagulation qui va limiter sa propre génération en activant ce système anticoagulant naturel. L’activation de la PC par la thrombine est une réaction extrêmement lente qui est considérablement accélérée (20 000 fois) en présence de thrombomoduline (TM), un récepteur endothélial qui agit comme un cofacteur dans ce système » (tiré de [57]).
L’agrégation plaquettaire est l’étape initiale de l’hémostase aboutissant au thrombus.
Références
- Kuhn JH, Bao Y, Bavari S, et al. Virus nomenclature below the species level: a standardized nomenclature for laboratory animal-adapted strains and variants of viruses assigned to the family Filoviridae. Arch Virol 2013 ; 158 : 1425–1432. [CrossRef] [PubMed] [Google Scholar]
- Sanchez A, Kiley MP, Holloway BP, Auperin DD. Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res 1993 ; 29 : 215–240. [CrossRef] [PubMed] [Google Scholar]
- Volchkov VE, Volchkova VA, Muhlberger E, et al. Recovery of infectious Ebola virus from complementary DNA: RNA editing of the GP gene and viral cytotoxicity. Science 2001 ; 291 : 1965–1969. [CrossRef] [PubMed] [Google Scholar]
- Reid SP, Leung LW, Hartman AL, et al. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol 2006 ; 80 : 5156–5167. [CrossRef] [PubMed] [Google Scholar]
- Valmas C, Grosch MN, Schumann M, et al. Marburg virus evades interferon responses by a mechanism distinct from Ebola virus. PLoS Pathog 2010 ; 6 : e1000721. [CrossRef] [PubMed] [Google Scholar]
- Page A, Volchkova VA, Reid SP, et al. Marburgvirus hijacks nrf2-dependent pathway by targeting nrf2-negative regulator keap1. Cell Rep 2014 ; 6 : 1026–1036. [CrossRef] [PubMed] [Google Scholar]
- Rouquet P, Froment JM, Bermejo M, et al. Wild animal mortality monitoring and human Ebola outbreaks, Gabon and Republic of Congo, 2001–2003. Emerg Infect Dis 2005 ; 11 : 283–290. [CrossRef] [PubMed] [Google Scholar]
- Swanepoel R, Leman PA, Burt FJ, et al. Experimental inoculation of plants and animals with Ebola virus. Emerg Infect Dis 1996 ; 2 : 321–325. [CrossRef] [PubMed] [Google Scholar]
- Leroy EM, Kumulungui B, Pourrut X, et al. Fruit bats as reservoirs of Ebola virus. Nature 2005 ; 438 : 575–576. [CrossRef] [PubMed] [Google Scholar]
- Towner JS, Amman BR, Sealy TK, et al. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog 2009 ; 5 : e1000536. [CrossRef] [PubMed] [Google Scholar]
- Negredo A, Palacios G, Vazquez-Moron S, et al. Discovery of an Ebola virus-like filovirus in europe. PLoS Pathog 2011 ; 7 : e1002304. [CrossRef] [PubMed] [Google Scholar]
- Geisbert TW, Hensley LE. Ebola virus: new insights into disease aetiopathology and possible therapeutic interventions. Expert Rev Mol Med 2004; 6 : 1–24. [CrossRef] [Google Scholar]
- Peters CJ, LeDuc JW. An introduction to Ebola: the virus and the disease. J Infect Dis 1999; 179 (suppl 1) : ix–xvi. [CrossRef] [PubMed] [Google Scholar]
- Baize S, Pannetier D, Oestereich L, et al. Emergence of Zaire Ebola virus disease in Guinea. N Engl J Med 2014 ; 371 : 1418–1425. [CrossRef] [PubMed] [Google Scholar]
- Aleksandrowicz P, Wolf K, Falzarano D, et al. Viral haemorrhagic fever and vascular alterations. Hamostaseologie 2008 ; 28 : 77–84. [PubMed] [Google Scholar]
- Baize S, Leroy EM, Georges AJ, et al. Inflammatory responses in Ebola virus-infected patients. Clin Exp Immunol 2002 ; 128 : 163–168. [CrossRef] [PubMed] [Google Scholar]
- Baize S, Leroy EM, Mavoungou E, Fisher-Hoch SP. Apoptosis in fatal Ebola infection. Does the virus toll the bell for immune system? Apoptosis 2000 ; 5 : 5–7. [CrossRef] [PubMed] [Google Scholar]
- Leroy EM, Baize S, Volchkov VE, et al. Human asymptomatic Ebola infection and strong inflammatory response. Lancet 2000 ; 355 : 2210–2215. [CrossRef] [PubMed] [Google Scholar]
- Geisbert TW, Hensley LE, Larsen T, et al. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am J Pathol 2003 ; 163 : 2347–2370. [CrossRef] [PubMed] [Google Scholar]
- Geisbert TW, Young HA, Jahrling PB, et al. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am J Pathol 2003 ; 163 : 2371–2382. [CrossRef] [PubMed] [Google Scholar]
- Mahanty S, Bray M. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect Dis 2004 ; 4 : 487–498. [CrossRef] [PubMed] [Google Scholar]
- Kortepeter MG, Bausch DG, Bray M. Basic clinical and laboratory features of filoviral hemorrhagic fever. J Infect Dis 2011 ; 204 : suppl 3 S810–S816. [CrossRef] [PubMed] [Google Scholar]
- Perry DL, Bollinger L, White GL. The baboon (Papio spp.) as a model of human Ebola virus infection. Viruses 2012; 4 : 2400–2416. [CrossRef] [PubMed] [Google Scholar]
- Zaki SR, Goldsmith CS. Pathologic features of filovirus infections in humans. Curr Top Microbiol Immunol 1999 ; 235 : 97–116. [PubMed] [Google Scholar]
- Zampieri CA, Sullivan NJ, Nabel GJ. Immunopathology of highly virulent pathogens: insights from Ebola virus. Nat immunol 2007 ; 8 : 1159–1164. [CrossRef] [PubMed] [Google Scholar]
- Wahl-Jensen VM, Afanasieva TA, Seebach J, et al. Effects of Ebola virus glycoproteins on endothelial cell activation and barrier function. J Virol 2005 ; 79 : 10442–10450. [CrossRef] [PubMed] [Google Scholar]
- Baize S, Leroy EM, Georges-Courbot MC, et al. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat Med 1999 ; 5 : 423–426. [CrossRef] [PubMed] [Google Scholar]
- Hoenen T, Groseth A, Falzarano D, Feldmann H. Ebola virus: unravelling pathogenesis to combat a deadly disease. Trends Mol Med 2006 ; 12 : 206–215. [CrossRef] [PubMed] [Google Scholar]
- Reynard OJ, Jacquot F, Volchkov V. L’infection à virus Ebola et les modèles animaux associés. Rev Fr Histotechnol 2012 ; 25 : 67–82. [Google Scholar]
- Geisbert TW, Young HA, Jahrling PB, et al. Mechanisms underlying coagulation abnormalities in Ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis 2003 ; 188 : 1618–1629. [CrossRef] [PubMed] [Google Scholar]
- Camerer E, Kolsto AB, Prydz H. Cell biology of tissue factor, the principal initiator of blood coagulation. Thromb Res 1996 ; 81 : 1–41. [CrossRef] [PubMed] [Google Scholar]
- Ernofsson M, Siegbahn A. Platelet-derived growth factor-BB and monocyte chemotactic protein-1 induce human peripheral blood monocytes to express tissue factor. Thromb Res 1996 ; 83 : 307–320. [CrossRef] [PubMed] [Google Scholar]
- Chen VM, Hogg PJ. Encryption and decryption of tissue factor. J Thromb Haemost 2013 ; 11 : suppl 1 277–284. [CrossRef] [PubMed] [Google Scholar]
- Rao LV, Pendurthi UR. Tissue factor-factor VIIa signaling. Arterioscler Thromb Vasc Biol 2005 ; 25 : 47–56. [PubMed] [Google Scholar]
- Geisbert TW, Hensley LE, Jahrling PB, et al. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet 2003 ; 362 : 1953–1958. [CrossRef] [PubMed] [Google Scholar]
- Hensley LE, StevensEL Yan SB, et al. Recombinant human activated protein C for the postexposure treatment of Ebola hemorrhagic fever. J Infect Dis 2007 ; 196 suppl 2 : S390–S399. [CrossRef] [PubMed] [Google Scholar]
- McElroy AK, Erickson BR, Flietstra TD, et al. Ebola hemorrhagic fever: novel biomarker correlates of clinical outcome. J Infect Dis 2014 ; 210 : 558–566. [CrossRef] [PubMed] [Google Scholar]
- Arata AA, Johnson B, Approaches towards studies on potential reservoirs of viral haemorrhagic fever in southern Sudan (1977). In : Pattyn SR, ed. Proceedings of an international colloquium on Ebola virus infection and other haemorrhagic fevers held in Antwerp, Belgium, 6–8 December, 1977. Amsterdam : Elsevier, 1978 : 136–142. [Google Scholar]
- Leroy EM, Baize S, Debre P, et al. Early immune responses accompanying human asymptomatic Ebola infections. Clin Exp Immunol 2001 ; 124 : 453–460. [CrossRef] [PubMed] [Google Scholar]
- Villinger F, Rollin PE, Brar SS, et al. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J Infect Dis 1999 ; 179 suppl 1 : S188–S191. [CrossRef] [PubMed] [Google Scholar]
- Gupta M, Mahanty S, Ahmed R, Rollin PE. Monocyte-derived human macrophages and peripheral blood mononuclear cells infected with Ebola virus secrete MIP-1alpha and TNF-alpha and inhibit poly-IC-induced IFN-alpha in vitro. Virology 2001 ; 284 : 20–25. [CrossRef] [PubMed] [Google Scholar]
- Ignatiev GM, Dadaeva AA, Luchko SV, Chepurnov AA. Immune and pathophysiological processes in baboons experimentally infected with Ebola virus adapted to guinea pigs. Immunol Lett 2000 ; 71 : 131–140. [CrossRef] [PubMed] [Google Scholar]
- Bosio CM, Moore BD, Warfield KL, et al. Ebola and Marburg virus-like particles activate human myeloid dendritic cells. Virology 2004 ; 326 : 280–287. [CrossRef] [PubMed] [Google Scholar]
- Ye L, Lin J, Sun Y, et al. Ebola virus-like particles produced in insect cells exhibit dendritic cell stimulating activity and induce neutralizing antibodies. Virology 2006 ; 351 : 260–270. [CrossRef] [PubMed] [Google Scholar]
- Jansen PM, Boermeester MA, Fischer E, et al. Contribution of interleukin-1 to activation of coagulation and fibrinolysis, neutrophil degranulation, and the release of secretory-type phospholipase A2 in sepsis: studies in nonhuman primates after interleukin-1 alpha administration and during lethal bacteremia. Blood 1995 ; 86 : 1027–1034. [PubMed] [Google Scholar]
- Stouthard JM, Levi M, Hack CE, et al. Interleukin-6 stimulates coagulation, not fibrinolysis, in humans. Thromb Haemost 1996 ; 76 : 738–742. [PubMed] [Google Scholar]
- Sanchez A, Lukwiya M, Bausch D, et al. Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: cellular responses, virus load, and nitric oxide levels. J Virol 2004 ; 78 : 10370–10377. [CrossRef] [PubMed] [Google Scholar]
- Tousoulis D, Kampoli AM, Tentolouris C, et al. The role of nitric oxide on endothelial function. Curr Vascular Pharmacol 2012 ; 10 : 4–18. [CrossRef] [Google Scholar]
- Martinez O, Valmas C, Basler CF. Ebola virus-like particle-induced activation of NF-kappaB and Erk signaling in human dendritic cells requires the glycoprotein mucin domain. Virology 2007 ; 364 : 342–354. [CrossRef] [PubMed] [Google Scholar]
- Escudero-Perez B, Volchkova VA, Dolnik O, et al. Shed GP of Ebola virus triggers immune activation and increased vascular permeability. PLoS Pathog 2014 ; 10 : e1004509. [CrossRef] [PubMed] [Google Scholar]
- Brudner M, Karpel M, Lear C, et al. Lectin-dependent enhancement of Ebola virus infection via soluble and transmembrane C-type lectin receptors. PLoS One 2013 ; 8 : e60838. [CrossRef] [PubMed] [Google Scholar]
- Krarup A, Wallis R, Presanis JS, et al. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS One 2007 ; 2 : e623. [CrossRef] [PubMed] [Google Scholar]
- Mayilyan KR, Presanis JS, Arnold JN, et al. Heterogeneity of MBL-MASP complexes. Mol Immunol 2006 ; 43 : 1286–1292. [CrossRef] [PubMed] [Google Scholar]
- Castro JE, Vado-Solis I, Perez-Osorio C, Fredeking TM. Modulation of cytokine and cytokine receptor/antagonist by treatment with doxycycline and tetracycline in patients with dengue fever. Clin Dev Immunol 2011 ; 2011 : 370872. [PubMed] [Google Scholar]
- Salgado D, Zabaleta TE, Hatch S, et al. Use of pentoxifylline in treatment of children with dengue hemorrhagic fever. Pediatr Infect Dis J 2012 ; 31 : 771–773. [CrossRef] [PubMed] [Google Scholar]
- Kerbiriou-Nabias D.. Les polyphosphates : nouveaux acteurs plaquettaires qui associent thrombose et inflammation. Med Sci (Paris) 2010 ; 26 : 343–346. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Borgel D, Vieillard-Baron A. La protéine C activée. Med Sci (Paris) 2011 ; 27 : 501–507. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Les chauves-souris, réservoirs présumés des filovirus. A. Hypsignathus monstrosus. B. Epomops franqueti. C. Myonycteris torquata. D. Rousettus aegyptiacus. E. Miniopterus Schreibersi. |
| Dans le texte | |
|
Figure 2. Description de la fièvre hémorragique à virus Ebola. Schéma du développement des fievres hémorragiques à virus Ebola à partir des données des épidemies de Mosango, de Yambuku et Gulu (données OMS et [21]). |
| Dans le texte | |
|
Figure 3. Rôle du facteur tissulaire dans l’initiation de la coagulation. L’association du facteur tissulaire (FT) avec le facteur VIIa constitue l’élément initiateur de nombreuses voies d’activation et notamment celle de l’activation du fibrinogène. |
| Dans le texte | |
|
Figure 4. Origine multifactorielle du dérèglement de l’hémostase dans l’infection à filovirus. L’infection virale conduit au dérèglement de nombreux mécanismes qui par synergie résulte en l’instauration d’une CIVD et d’un état de choc. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.