")
")
| Issue |
Med Sci (Paris)
Volume 30, Number 12, Décembre 2014
|
|
|---|---|---|
| Page(s) | 1177 - 1183 | |
| Section | Forum | |
| DOI | https://doi.org/10.1051/medsci/20143012022 | |
| Published online | 24 December 2014 | |
Syllogomanie moléculaire : l’ADN non codant enrichit le jeu des possibles
Compulsive molecular hoarding enables the evolution of protein-coding DNA from non-coding DNA
Laboratoire évolution, génome et spéciation, UPR 9034 CNRS, avenue de la Terrasse, 91198
Gif-sur-Yvette, France
université Paris-Diderot, UFR des sciences du vivant, Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Résumé
Il était admis, jusqu’à très récemment, qu’un nouveau gène codant une nouvelle protéine ne pouvait avoir comme origine qu’un gène préexistant, une combinaison de fragments de gènes, ou un transfert horizontal de gène à partir d’une autre espèce. Une série d’études comparatives de génomes et de transcriptomes suggèrent qu’il existe une autre source de gènes codant des protéines : l’ADN non codant. Le mécanisme, vraisemblablement universel car proposé pour divers groupes d’eucaryotes, implique l’existence d’un continuum de protogènes entre ADN codant et non codant, correspondant à des états intermédiaires fixés par la sélection naturelle. Ainsi, au cœur des génomes, des gènes pourraient émerger progressivement du néant par le seul jeu des mutations et de la sélection naturelle.
Abstract
It was thought until recently that a new gene could only evolve from a previously existing gene, from recombination of genes, or from horizontal gene transfer. Recently a series of genomic and transcriptomic studies have led to the identification of non-coding DNA as a significant source of protein coding genes. The mechanism, which is probably universal since it has been identified in a wide array of eukaryotes, implies that a gradient of proto-genes, probably established by a balance between selection and genetic drift, exists between coding DNA and non-coding DNA. Therefore genome dynamics could account for the progressive formation of genes “out of the blue” thanks to the interplay of mutation and natural selection.
© 2014 médecine/sciences – Inserm

Les quelques phrases (Encadré 1) extraites du livre de François Jacob « Le jeu des possibles, essai sur la diversité du vivant » [1], publié en 1981, illustrent parfaitement le cadre théorique dans lequel fut étudié, jusqu’à très récemment, l’évolution du contenu des génomes en gènes codant des protéines.
1
« L’évolution ne tire pas ses nouveautés du néant. Elle travaille sur ce qui existe déjà, soit qu’elle transforme un système ancien pour lui donner une fonction nouvelle, soit qu’elle combine plusieurs systèmes pour en échafauder un autre plus complexe […]
La probabilité de voir une protéine fonctionnelle se former de novo, par association au hasard d’acides aminés, est pratiquement zéro. Chez des organismes aussi complexes et intégrés que ceux ayant vécu il y a déjà fort longtemps, la création de séquences nucléiques entièrement nouvelles ne pouvait jouer un rôle important dans la production d’information nouvelle. Durant la majeure partie de l’évolution biologique, la création de structures moléculaires ne pouvait se fonder que sur un remaniement de structures préexistantes […]
Très vraisemblablement, tout a commencé avec de petites séquences de 30 à 50 nucléotides produites par l’évolution chimique et capables chacune de coder de 10 à 15 acides aminés. C’est seulement après coup que de telles séquences ont dû être unies au hasard par des processus de ligature pour former des chaînes protéiques plus longues. Certaines de celles-ci se sont avérées utiles et ont été sélectionnées […]
Une fois encore, on voit mal comment l’évolution moléculaire aurait pu procéder si ce n’est en faisant du neuf avec du vieux ; en liant ensemble des morceaux d’ADN ; bref en bricolant. »
François Jacob
Le jeu des possibles, essai sur la diversité du vivant (1981)
L’argumentation repose sur deux hypothèses : (1) les premières étapes de la formation du vivant permettaient l’émergence de novo de courtes séquences codant des protéines, ce qui n’est plus le cas aujourd’hui ; un principe qu’on pourrait qualifier de « non actualisme » ; et (2) après cette première étape, l’évolution a consisté à faire du neuf (des gènes codant de nouvelles protéines) avec du vieux (des gènes déjà existants) ; c’est le concept de bricolage évolutif (genetic tinkering), cher à François Jacob [2]. Bien sûr, et comme presque toujours en biologie évolutive, quelques contre-exemples furent identifiés, mais ils furent considérés comme des exceptions qui confirmaient la règle [3, 4]. Cependant, quelques publications récentes remettent en cause ce paradigme et proposent que, chez les eucaryotes, une proportion importante, de 5 % à 12 %, des gènes codant des protéines spécifiques de quelques organismes, voire d’une seule espèce, pourraient avoir l’ADN non codant comme origine [5, 6]. Entre les deux catégories, ADN non codant (non gène) et ADN codant (gène), il existerait un continuum de protogènes correspondant à des états intermédiaires entre ces deux états limites [7]. Dans le cadre de cette revue, nous ne nous intéresserons qu’à l’origine des gènes codant des protéines, car il existe une grande quantité de gènes transcrits en des ARN qui ne sont pas traduits en protéines, et dont l’origine de novo est très vraisemblable. Nous verrons toutefois qu’il existe probablement un lien étroit entre ces deux types de gènes [8].
Bricolage moléculaire
Pendant plusieurs décennies, il fut accepté que seuls trois mécanismes pouvaient expliquer l’apparition de nouveaux gènes dans un génome. Le plus simple, et le premier identifié, consiste en la duplication d’un gène par enjambement inégal (unequal crossing-over), qui produit deux copies filles à partir d’un gène préexistant [9, 10]. À l’origine identiques, ces copies accumulent au cours de l’évolution des mutations qui entraînent peu à peu la divergence des séquences des protéines codées. Il est à noter qu’après une duplication, très rapidement, les protéines codées peuvent être très différentes s’il se produit un décalage du cadre de lecture dans un des gènes [11]. Dès 1936, Muller affirme « There remains no reason to doubt the application of the dictum “all life from pre-existing life” and “every cell from a pre-existing cell,” to the gene: “every gene from a pre-existing gene” » [12]. Identifiées dans les années 1920, les duplications de gène ont été considérées dans les années 1970 comme le principal mécanisme qui permet d’agrandir le répertoire des protéines codées par un génome [13, 14]. Il est ensuite apparu que des gènes sont aussi produits par recombinaison des séquences de plusieurs gènes (dont le mécanisme de brassage d’exons ou exon shuffling) [3, 4, 15]. Enfin, un génome peut acquérir de nouveaux gènes par transferts horizontaux, c’est-à-dire par intégration de fragments d’ADN provenant d’autres espèces, parfois très éloignées du point de vue phylogénétique [3, 4, 16–18, 52]. Ces différents mécanismes sont schématisés dans la Figure 1A–C .
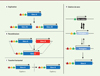 |
Figure 1. Les différents modes de formation de nouveaux gènes. A-C. À partir de gènes préexistants. A. Duplication à l’occasion d’une recombinaison non homologue (duplication en cis, dite duplication en tandem) ou d’une duplication totale du génome (duplication en trans) : un gène est dupliqué et forme deux copies filles, ou paralogues, qui sont à l’origine fonctionnellement redondantes. B. Recombinaison, à l’occasion d’une recombinaison non homologue ou d’une rétrotransposition : des parties codantes provenant de gènes différents fusionnent pour constituer un gène qui peut coder une protéine avec une nouvelle fonction. C. Transfert horizontal : transfert d’un gène d’une espèce à une autre ; ce mécanisme, très fréquent chez les bactéries, se produit également chez des eucaryotes, souvent en relation avec la phagocytose et le parasitisme qui rapprochent les génomes d’espèces très différentes. D. Création de novo : dans les génomes, il existe de très nombreuses séquences non codantes transcrites et qui contiennent un court cadre de lecture ouvert (ORF). Ces séquences, à l’origine non fonctionnelles, sont le plus souvent éliminées, car il n’y a pas de pression de sélection pour leur maintien. Elles sont aussi souvent associées à des séquences régulatrices qui contrôlent leur expression dans le temps et/ou l’espace. Si la protéine codée permet l’augmentation, aussi faible soit-elle, de la valeur sélective, le protogène peut être sélectionné et peut accumuler progressivement des modifications de la séquence codante et des séquences régulatrices jusqu’à former un nouveau gène qui aura une faible probabilité de disparaître au cours de l’évolution. Les séquences codantes sont figurées par des boîtes colorées, l’origine de transcription par une flèche brisée et les séquences régulatrices par des triangles de couleur (modifié d’après Carvunis et al. [7]). |
Les gènes orphelins
À la suite du séquençage total du génome de la levure en 1996, Bernard Dujon remarqua l’abondance inattendue de gènes dépourvus d’homologues et baptisa « orphelins » ces gènes sans gène apparenté connu [19]. Toutes les études de la composition en gènes des génomes effectuées depuis le confirment. Tant chez les eucaryotes [20] que chez les virus et les bactéries [21–26], les génomes portent toujours une part importante de gènes pour lesquels il ne peut être identifié ni gène paralogue (gène homologue issu d’une duplication de gène et pouvant se trouver dans une même espèce ou dans des espèces différentes), ni gène orthologue (gène homologue issu d’une spéciation et ne pouvant donc se trouver que dans des espèces différentes). Au départ, deux explications furent préférées à la création de gènes de novo (sans implication de séquences codantes préexistantes) : soit ces gènes orphelins avaient des séquences divergentes à un point tel qu’il n’était plus possible de reconnaître leur homologie avec d’autres séquences, soit leurs homologues n’avaient pas encore été identifiés chez d’autres espèces. Il est vrai que, pour des génomes d’eucaryotes contenant beaucoup d’ADN non codant et dans lequel des gènes orphelins furent très activement recherchés (comme les génomes des rongeurs et des primates qui sont parmi les mieux séquencés et les mieux annotés), il a longtemps subsisté un nombre d’erreurs assez important pour mettre en doute une part non négligeable des gènes identifiés comme orphelins [27]. Cependant, l’accumulation récente de données de séquençage total des génomes, dont notamment des génomes d’espèces phylogénétiquement très proches, a permis de lever ces doutes qui apparaissent de moins en moins fondés. Aussi, une explication alternative s’impose peu à peu : ces gènes seraient d’origine très récente et n’auraient pas été produits par l’un des mécanismes de génération de nouveaux gènes classiques (duplication, recombinaison, ou transfert horizontal)
Il existe donc des gènes qui ne sont présents que chez un groupe d’espèces étroitement apparentées (voire une seule espèce), ce qui permet de dater leur origine, entre l’âge du dernier ancêtre commun de ce groupe d’espèces et le moment de séparation avec la lignée qui est la plus proche parente de ce groupe (Figure 2). Cette approche, dite phylostratigraphique, permet d’identifier à quel moment est apparu un gène dans la lignée évolutive menant à l’espèce étudiée, en appliquant une approche phylogénétique que nous avons décrite précédemment [28, 29]. Une phylostratigraphie récente [30] montre que parmi les 22 773 gènes codants chez la souris, 7 253 gènes sont partagés avec les archées et les bactéries, et étaient donc présents chez LUCA (le dernier ancêtre commun universel) ; 8 438 gènes sont apparus dans la lignée de l’ancêtre des animaux depuis sa séparation d’avec LUCA ; 4 118 gènes sont apparus dans la lignée de l’ancêtre des vertébrés depuis sa séparation d’avec celle des éponges ; 1 382 gènes sont apparus dans la lignée de l’ancêtre des mammifères depuis sa séparation d’avec la lignée des requins ; 801 gènes sont apparus dans la lignée de l’ancêtre des rongeurs depuis sa séparation d’avec les marsupiaux et 781 n’existent que chez la souris (Figure 2). La plupart des gènes de la souris sont donc très anciens, environ 70 % d’entre eux étant apparus il y a plus de 550 millions d’années, c’est-à-dire avant la diversification des animaux. À l’inverse et de façon inattendue, 781 gènes semblent très récents, c’est-à-dire qu’il ne peut pas être identifié de séquence assez ressemblante pour la considérer comme homologue, même chez une espèce très proche comme le rat. Ces gènes seraient donc apparus de novo dans la lignée de la souris, après sa séparation de la lignée du rat. Les estimations de la date de séparation du rat et de la souris sont très variables selon les études, mais ces espèces auraient divergé il y a 10 à 25 millions d’années. Il semblerait donc que plusieurs centaines de gènes sont apparus dans cet intervalle de temps. En utilisant cette même approche, des nombres similaires de nouveaux gènes furent identifiés chez l’homme, la drosophile, un certain nombre de plantes et la levure du boulanger [7]. Il est possible qu’une large fraction de ces nouveaux gènes codants ne soient pas des gènes, c’est-à-dire qu’ils ne codent pas réellement des protéines et qu’ils ne soient que des erreurs d’annotation. Il ne faut pas perdre de vue que toutes ces études se fondent sur des annotations automatiques des génomes. En particulier, l’identification des séquences codantes des gènes est faite à partir de la détection d’ORF (open reading frame ou cadre ouvert de lecture), de données sur leur transcription en ARNm, de l’existence de protéines correspondantes et de données expérimentales qui indiquent que ces séquences ont un rôle fonctionnel au niveau de l’organisme. Très souvent, toutes ces données ne sont pas disponibles pour un gène donné. Des séquences peuvent être annotées comme codantes alors qu’elles ne le sont pas, et d’autres séquences peuvent ne pas être considérées comme codantes alors qu’elles le sont. Ainsi, au cours du temps, on peut voir des séquences codantes apparaître ou disparaître de l’annotation des génomes au gré des informations qui s’accumulent. De plus, l’identification d’une séquence codante chez une espèce est le plus souvent réalisée à partir de sa ressemblance avec une séquence codante d’une autre espèce. Il est donc très probable qu’une fraction importante des gènes codants n’ait pas encore été identifiée, même chez les espèces modèles. Le séquençage direct des protéines est une voie prometteuse pour identifier de façon plus complète l’ensemble des protéines codées par un génome et, ainsi, mettre en évidence de nouveaux gènes codants, en particulier ceux qui codent des protéines de petite taille [31, 32].
 |
Figure 2. Phylostratigraphie des gènes de la souris. Les gènes présents dans le génome de la souris sont comparés aux gènes présents dans les génomes d’autres espèces afin d’identifier le moment de leur apparition. En s’appuyant sur la phylogénie des espèces, l’âge d’un gène est déterminé en identifiant l’ensemble des espèces qui partagent ce gène avec la souris. Par exemple, si un gène n’existe que chez la souris, il est apparu après la séparation de l’ancêtre du rat et de celui de la souris. Si un gène est partagé uniquement par la souris et les autres mammifères, il est apparu après la séparation des mammifères de toutes les autres espèces et avant le dernier ancêtre commun des mammifères. Si un gène est présent chez la souris et toutes les autres espèces, il était donc déjà présent chez LUCA, l’ancêtre commun de toutes les espèces. AC : ancêtre commun (modifié d’après Neme et al. [30]). |
Par ailleurs, beaucoup de gènes apparentés (entièrement ou pour des domaines partagés par recombinaisons) et très anciens ne montrent aucune ressemblance entre eux, car leur évolution indépendante pendant des centaines de millions d’années, voire quelques milliards d’années, fait qu’il n’est plus possible d’identifier cette homologie sur la seule base de la comparaison de leurs séquences. Le nombre de familles de gènes homologues pourrait donc être une forte surévaluation du nombre de gènes apparus indépendamment.
L’analyse de l’ensemble des études récentes, en tenant compte des biais et imperfections des données énoncés ci-dessus, confirme néanmoins l’identification d’un nombre important de gènes codant des protéines formés de novo à partir d’ADN non codant [6].
La formation de gènes de novo
Jusqu’à très récemment, l’existence de gènes codants issus de l’ADN non codant était donc acceptée comme une curiosité [3, 4, 6], sans importance majeure en tant que mécanisme de création de nouvelles protéines. La possibilité de séquencer les génomes et les transcriptomes de plusieurs espèces choisies en fonction de critères phylogénétiques a permis de réévaluer l’importance de ce mécanisme.
Comme nous l’avons vu précédemment, l’identification de gènes potentiellement apparus de novo se fait en deux étapes : (1) une étape d’analyse informatique basée sur une approche comparative, la phylostratigraphie, qui permet d’estimer l’âge des gènes, les gènes orphelins les plus récents étant plus vraisemblablement apparus de novo ; et (2) une étape de validation expérimentale, l’analyse fonctionnelle, qui permet de vérifier que ces gènes codent réellement des protéines, et que celles-ci ont un effet sur la valeur sélective (fitness) de l’organisme. Cette seconde étape n’a encore été réalisée que dans de rares cas, et une validation de la fonction de la plupart des gènes issus de l’ADN non codant est encore à faire [33–35]. Dans quelques cas, on peut tester si la sélection naturelle a été impliquée dans la fixation d’un gène apparu de novo [36]. À ce jour, il est très difficile d’estimer quelle est la contribution des gènes apparus de novo à l’ensemble des gènes codants que l’on peut observer dans un génome. Il est toutefois très probable que cette part est plus grande qu’on ne le pensait jusqu’à récemment, et ces gènes pourraient constituer de 5 à 12 % des gènes apparus récemment chez diverses espèces [6].
Natura non facit saltum
Il est tout à fait invraisemblable qu’un gène codant une protéine, voire souvent plusieurs protéines grâce au mécanisme de l’épissage alternatif, puisse apparaître en une seule étape à partir d’une séquence non codante, au moins dans le cas d’un gène tel qu’on le définit habituellement, c’est-à-dire une séquence d’ADN (continue ou fragmentée si, respectivement, elle ne contient pas d’introns ou si elle contient des introns) qui présentent plusieurs caractéristiques : (1) elle est transcrite en au moins un ARNm, lui-même traduit car il contient au moins un cadre ouvert de lecture ; (2) elle est associée à un ensemble de séquences régulatrices permettant un contrôle fin de son expression ; (3) elle permet la synthèse d’au moins une protéine avec une ou plusieurs activités biologiques intégrées dans un système fonctionnel ayant un effet sur la valeur sélective de l’organisme. Pour que toutes ces propriétés structurales et fonctionnelles soient présentes, il faut donc supposer des états successifs, constitués par des protogènes, dont la fonction est d’abord mineure et mal intégrée, mais suffisamment utile pour que démarre le processus de sélection qui permet d’accumuler progressivement des mutations et, ainsi, d’améliorer le gène, tant au niveau de son expression que de la fonction de la protéine codée. Il faut donc imaginer des états intermédiaires entre l’ADN non codant et le gène codant une protéine (Figure 1D). Les protogènes les plus récents seront les moins optimisés et pourront aisément être éliminés. Les plus anciens seront plus difficilement éliminés, car ils correspondront davantage aux critères énoncés ci-dessus qui définissent un gène et seront donc plus souvent indispensables [37]. Mais ne perdons pas de vue que même des gènes très anciens et très conservés sont parfois perdus sporadiquement dans différentes lignées phylogénétiques [38]. Ce modèle de construction progressive des gènes à partir de rien implique que les premiers stades des protogènes sont très facilement atteints. C’est très vraisemblable, car l’ADN non codant est présent en très grande quantité dans le génome de beaucoup d’eucaryotes où souvent il représente plus de 90 % de l’ADN total [39–41]. La quasi-totalité du génome est transcrit en ARN [42] et beaucoup de ces ARN contiennent un ORF de petite taille [7]. Un ORF de petite taille, transcrit et codant une petite protéine, peut donc apparaître par hasard avec une fréquence non négligeable dans la grande quantité d’ADN non codant qui constitue l’essentiel du génome de la plupart des eucaryotes. Les génomes des bactéries et des virus sont généralement beaucoup moins riches en ADN non codant ; cela offre moins de chance de voir se former des séquences codantes au hasard dans un génome donné. Mais ces organismes forment des populations de bien plus grande taille que les eucaryotes. Ceci compense, en terme de quantité d’ADN, la faible teneur de ces génomes en ADN non codant et ouvre la possibilité théorique que bactéries et virus soient également capables de produire des gènes de novo. Chez les bactéries et les eucaryotes, les approches génomique, transcriptomique et protéomique ont permis de détecter la transcription de nombreux petits ARNm, certains codant effectivement de petites protéines dont les fonctions sont, pour la plupart, inconnues [31]. Si un gène apparu de novo permet la synthèse d’une nouvelle protéine et si celle-ci se révèle utile à l’organisme, et augmente un tant soit peu sa valeur sélective, il peut s’enclencher un processus de mutation/sélection qui optimise pas à pas la fonction de cette protéine par la fixation de nouvelles mutations au cours du temps, tant au niveau de la séquence codante que des séquences régulatrices (Figure 1D). Il est donc attendu dans le cadre de ce modèle que plus les gènes de novo sont anciens, plus ils sont complexes, c’est-à-dire qu’ils sont plus grands, que leur régulation fait intervenir plus de séquences régulatrices, qu’ils contiennent plus d’introns (qui ont eu plus de temps pour s’insérer dans la séquence codante unique de départ), et qu’ils font un usage plus optimisé des codons. Des études réalisées chez l’homme, la drosophile et la levure du boulanger montrent que l’ensemble de ces attendus sont effectivement observés [7, 30, 37].
Conclusions
Si la plupart des gènes codant pour protéines se forment bien dans le cadre du paradigme du genetic tinkering posé par François Jacob il y a plusieurs décennies, il apparaît qu’une fraction non négligeable (probablement de l’ordre de 10 %) de nouveaux gènes émergeraient de novo à partir d’ADN non codant. Au-delà de l’identification d’un nouveau mode de formation de nouveaux gènes, ce mécanisme éclaire de façon intéressante deux problèmes de biologie évolutive de portée très générale.
Syllogomanie moléculaire
Le génome de la plupart des eucaryotes est très riche en ADN non codant. Chez les plantes et les animaux, celui-ci constitue souvent l’essentiel du génome [39, 40]. Or, il existe des exemples de plantes et d’animaux dont les génomes ont perdu une grande partie de leur ADN non codant [43, 44], ou qui l’éliminent dans les lignées somatiques [45, 46]. Ceci montre que la plus grande partie de l’ADN non codant n’est pas indispensable à la survie et/ou la reproduction des individus [41, 47]. Ainsi, seulement 5 % du génome humain seraient soumis à sélection [48] et donc fonctionnel d’un point de vue évolutif. Le maintien, chez la plupart des espèces eucaryotes, de cette énorme quantité d’ADN non codant et non fonctionnel pourrait simplement montrer les limites de l’efficacité de la sélection naturelle. En effet, si la variation de la quantité d’ADN non codant ne se traduit pas par une variation assez importante de la valeur sélective des individus, la sélection naturelle n’est pas assez efficace pour éliminer du génome cet ADN « inutile » [49]. Il est alors frappant de constater que de cette énorme quantité d’ADN « poubelle » (junk DNA), réputé n’avoir aucune fonction, pourrait être à l’origine, non seulement de séquences d’ADN et de transcrits ARN non codants aux multiples fonctions régulatrices, mais aussi du codage de nouvelles protéines. Non contente d’être une bricoleuse qui assemble des fragments de gènes disparates pour créer de nouveaux gènes, l’évolution se comporte aussi comme une syllogomane qui ne rechigne pas à fouiller dans les poubelles du génome !
Origine des structures complexes
Les adversaires de la théorie de l’évolution soulignent à l’envi qu’il est fort improbable qu’une structure biologique complexe puisse apparaître « à partir du néant ». Suivant en cela les préceptes du théologien William Paley, ils voient dans cette complexité la preuve de l’existence d’une volonté, d’un dessein intelligent. Pour eux, en somme, pas d’horloge sans « Grand horloger » ! C’est méconnaître la puissance du couple mutation/sélection dans la constitution d’une structure très complexe à partir d’une structure très simple, par accumulation de petits changements successifs sur de grandes périodes de temps et ce, sans implication d’évènements hautement improbables (Figure 3). Si aucun objet complexe ne peut être produit instantanément et par hasard, il existe beaucoup d’exemples morpho-anatomiques qui démontrent comment, par petites étapes successives, l’apparition continuelle de modifications aléatoires suivies d’un tri par la sélection naturelle aboutit, sur une longue période de temps, à la mise en place progressive de structures très complexes. L’existence, en quantité non négligeable, de gènes codants issus de l’ADN non codant étend jusqu’au cœur même des génomes l’implication du couple mutation/sélection dans la formation ex-nihilo de structures complexes. Ce qui frappe dans ce phénomène, comme pour tout mécanisme d’essai/erreur, c’est l’énorme gaspillage induit par ce processus. En effet, cela implique non seulement qu’une partie importante de l’activité de transcription et de traduction est consacrée à produire des molécules peu ou pas fonctionnelles, mais aussi que la plupart de celles-ci seront éliminées. Comment un tel coût métabolique est-il supporté ? S’agit-il d’un simple mécanisme de « fuite » dû au coût métabolique trop élevé d’un contrôle plus précis de la transcription ? Est-ce le résultat du fait que la sélection ne peut optimiser un système biologique au-delà d’un certain niveau dans une population de taille finie [50, 51], ou s’agit-il d’un mécanisme sélectionné qui témoigne d’un compromis entre optimisation et évolvabilité des génomes ? La question reste ouverte.
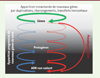 |
Figure 3. Les deux voies de formation des nouveaux gènes. La plupart des nouveaux gènes qui apparaissent au sein des génomes sont formés par duplication, fusion ou transfert horizontal (boucle verte). Cependant, 5 à 12 % des gènes codant des protéines seraient formés de novo à partir d’ADN non codant. Ces nouveaux gènes apparaitraient progressivement ; des protogènes de plus en plus conformes aux propriétés des gènes constitueraient un gradient entre les deux états non-gène/gène. Cette création de gènes de novo serait contrebalancée par l’élimination continue de gènes (pseudogénisation) et de protogènes (modifié d’après Carvunis et al. [7]). |
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Nous tenons à exprimer ici toute notre gratitude à nos collègues Mélanie Debiais-Thibaud et Alice Michel-Salzat pour leur relecture attentive et critique de notre manuscrit, ainsi qu’à notre collègue Cushla Metcalfe, pour l’amélioration des titres et résumé en anglais.
Références
- Jacob F. Le Jeu des possibles, essai sur la diversité du vivant. Paris : Fayard, 1981. [Google Scholar]
- Jacob F. Evolution and tinkering. Science 1977 ; 196 : 1161–1166. [CrossRef] [PubMed] [Google Scholar]
- Long M, Betran E, Thornton K, Wang W. The origin of new genes: glimpses from the young and old. Nat Rev Genet 2003 ; 4 : 865–875. [CrossRef] [PubMed] [Google Scholar]
- Long M, VanKuren NW, Chen S, Vibranovski MD. New gene evolution: little did we know. Annu Rev Genet 2013 ; 47 : 307–333. [CrossRef] [PubMed] [Google Scholar]
- Tautz D, Domazet-Lošo T. The evolutionary origin of orphan genes. Nat Rev Genet 2011 ; 12 : 692–702. [CrossRef] [PubMed] [Google Scholar]
- Ding Y, Zhou Q, Wang W. Origins of new genes and evolution of their novel functions. Annu Rev Ecol Evol Syst 2012 ; 43 : 345–363. [CrossRef] [Google Scholar]
- Carvunis AR, Rolland T, Wapinski I, et al. Protogenes and de novo gene birth. Nature 2012 ; 487 : 370–374. [CrossRef] [PubMed] [Google Scholar]
- Xie C, Zhang YE, Chen JY, et al. Hominoid-specific de novo protein-coding genes originating from long non-coding RNAs. PLoS Genet 2012 ; 8 : e1002942. [CrossRef] [PubMed] [Google Scholar]
- Bridges CB. The bar “gene” - A duplication. Science 1936 ; 83 : 210–211. [CrossRef] [PubMed] [Google Scholar]
- Sturtevant AH. The effects of unequal crossing over at the bar locus in Drosophila. Genetics 1925 ; 10 : 117–147. [PubMed] [Google Scholar]
- Ohno S. Birth of a unique enzyme from an alternative reading frame of the preexisted, internally repetitious coding sequence. Proc Natl Acad Sci USA 1984 ; 81 : 2421–2425. [CrossRef] [Google Scholar]
- Muller HJ. Bar duplication. Science 1936 ; 83 : 528–530. [CrossRef] [PubMed] [Google Scholar]
- Miquelis A, Abi-Rached L, Gilles A, Pontarotti P. Mise en évidence de processus de duplications en bloc dans le génome des vertébrés. Med Sci (Paris) 2002 ; 18 : 1051–1054. [CrossRef] [EDP Sciences] [Google Scholar]
- Ohno S., Evolution by gene duplication. Berlin : Springer-Verlag, 1970. [CrossRef] [Google Scholar]
- Gilbert W. Why genes in pieces? Nature 1978 ; 271 : 501. [CrossRef] [PubMed] [Google Scholar]
- Daubin V, Abby S. Les transferts horizontaux de gènes et l’arbre de la vie. Med Sci (Paris) 2012 ; 28 : 695–698. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Da Lage JL, Binder M, Hua-Van A, et al. Gene make-up: rapid, massive intron gains after horizontal transfer of a bacterial alpha-amylase gene to Basidiomycetes. BMC Evol Biol 2013 ; 13 : 40. [CrossRef] [PubMed] [Google Scholar]
- Da Lage JL, Danchin EG, Casane D. Where do animal alpha-amylases come from? An interkingdom trip. FEBS Lett 2007 ; 581 : 3927–3935. [CrossRef] [PubMed] [Google Scholar]
- Dujon B. The yeast genome project: What did we learn?. Trends Genet 1996 ; 12 : 263–270. [CrossRef] [PubMed] [Google Scholar]
- Khalturin K, Hemmrich G, Fraune S, et al. More than just orphans: are taxonomically-restricted genes important in evolution? Trends Genet 2009 ; 25 : 404–413. [CrossRef] [PubMed] [Google Scholar]
- Daubin V, Ochman H. Bacterial genomes as new gene homes: The genealogy of ORFans in E. coli. Genome Res 2004 ; 14 : 1036–1042. [CrossRef] [PubMed] [Google Scholar]
- Fischer D, Eisenberg D. Finding families for genomic ORFans. Bioinformatics 1999 ; 15 : 759–762. [CrossRef] [PubMed] [Google Scholar]
- Pavesi A, Magiorkinis G, Karlin DG., Viral proteins originated de novo by overprinting can be identified by codon usage: Application to the “gene nursery” of deltaretroviruses. PLoS Comput Biol 2013 ; 9 : e1003162. [CrossRef] [PubMed] [Google Scholar]
- Rancurel C, Khosravi M, Dunker AK, et al. Overlapping genes produce proteins with unusual Sequence properties and offer insight into de novo protein creation. J Virol 2009 ; 83 : 10719–10736. [CrossRef] [PubMed] [Google Scholar]
- Yin Y, Fischer D. Identification investigation of ORFans in the viral world. BMC Genomics 2008 ; 9 : 24. [CrossRef] [PubMed] [Google Scholar]
- Yin YB, Fischer D., On the origin of microbial ORFans: quantifying the strength of the evidence for viral lateral transfer. BMC Evol Biol 2006 ; 6 : 63. [CrossRef] [PubMed] [Google Scholar]
- Murphy DN, McLysaght A., De novo origin of protein-coding genes in murine rodents. PLoS One 2012 ; 7 : e48650. [CrossRef] [PubMed] [Google Scholar]
- Casane D, Laurenti P. Une toute nouvelle tête pour l’ancêtre des vertébrés à mâchoires. Med Sci (Paris) 2014 ; 30 : 38–40. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Casane D, Laurenti P. Penser la biologie dans un cadre phylogénétique. L’exemple de l’évolution des vertébrés. Med Sci (Paris) 2012 ; 28 : 1121–1127. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Neme R, Tautz D., Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC Genomics 2013 ; 14 : 117. [CrossRef] [PubMed] [Google Scholar]
- Andrews SJ, Rothnagel JA. Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet 2014 ; 15 : 193–204. [CrossRef] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, et al. A draft map of the human proteome. Nature 2014 ; 509 : 575–581. [CrossRef] [PubMed] [Google Scholar]
- Heinen TJAJ, Staubach F, Häming D, Tautz D. Emergence of a new gene from an intergenic region. Curr Biol 2009 ; 19 : 1527–1531. [CrossRef] [PubMed] [Google Scholar]
- Ranz JM, Parsch J. Newly evolved genes: Moving from comparative genomics to functional studies in model systems. Bioessays 2012 ; 34 : 477–483. [CrossRef] [PubMed] [Google Scholar]
- Reinhardt JA, Wanjiru BM, Brant AT, et al. De novo ORFs in Drosophila are important to organismal fitness, evolved rapidly from previously non-coding sequences. PLoS Genet 2013 ; 9 : e1003860. [CrossRef] [PubMed] [Google Scholar]
- Zhao L, Saelao P, Jones CD, Begun DJ. Origin and spread of de novo genes in Drosophila melanogaster populations. Science 2014 ; 343 : 769–772. [CrossRef] [PubMed] [Google Scholar]
- Palmieri N, Kosiol C, Schlotterer C., The life cycle of Drosophila orphan genes. Elife 2014 ; 3 : e01311. [CrossRef] [PubMed] [Google Scholar]
- Blomme T, Vandepoele K, De Bodt S, et al. The gain, loss of genes during 600 million years of vertebrate evolution. Genome Biol 2006 ; 7 : R43. [CrossRef] [PubMed] [Google Scholar]
- Metcalfe CJ, Casane D., Accommodating the load: The transposable element content of very large genomes. Mob Genet Elements 2013 ; 3 : e24775. [CrossRef] [PubMed] [Google Scholar]
- Metcalfe CJ, Filee J, Germon I, et al. Evolution of the Australian lungfish (Neoceratodus forsteri) genome: a major role for CR1 and L2 LINE elements. Mol Biol Evol 2012 ; 29 : 3529–3539. [CrossRef] [PubMed] [Google Scholar]
- Palazzo AF, The Gregory TR., case for Junk DNA., Genet PLoS, ; 2014 ; 10 : e1004351. [Google Scholar]
- Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol 2007 ; 14 : 103–105. [CrossRef] [PubMed] [Google Scholar]
- Ibarra-Laclette E, Lyons E, Hernandez-Guzman G, et al. Architecture and evolution of a minute plant genome. Nature 2013 ; 498 : 94–98. [CrossRef] [PubMed] [Google Scholar]
- Aparicio S, Chapman J, Stupka E, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 2002 ; 297 : 1301–1310. [CrossRef] [PubMed] [Google Scholar]
- Sun C, Wyngaard G, Walton D, et al. Billions of basepairs of recently expanded, repetitive sequences are eliminated from the somatic genome during copepod development. BMC Genomics 2014 ; 15 : 186. [CrossRef] [PubMed] [Google Scholar]
- Smith JJ, Antonacci F, Eichler EE, Amemiya CT. Programmed loss of millions of base pairs from a vertebrate genome. Proc Natl Acad Sci USA 2009 ; 106 : 11212–11217. [CrossRef] [Google Scholar]
- Doolittle WF, Brunet TDP, Linquist S, Gregory TR. Distinguishing between “function” and “effect” in genome biology. Genome Biol Evol 2014 ; 6 : 1234–1237. [CrossRef] [PubMed] [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011 ; 478 : 476–482. [CrossRef] [PubMed] [Google Scholar]
- Lynch M. The origins of eukaryotic gene structure. Mol Biol Evol 2006 ; 23 : 450–468. [CrossRef] [PubMed] [Google Scholar]
- Lynch M., The origins of genome architecture, Sunderland, Massachusetts: Sinauer, 2007. [Google Scholar]
- Lynch M. Evolution of the mutation rate. Trends Genet 2010 ; 26 : 345–352. [CrossRef] [PubMed] [Google Scholar]
- Gilbert C, Schaack S, Feschotte C. Quand les éléments génétiques mobiles bondissent entre espèces animales. Med Sci (Paris) 2010 ; 26 : 1025–1027. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Les différents modes de formation de nouveaux gènes. A-C. À partir de gènes préexistants. A. Duplication à l’occasion d’une recombinaison non homologue (duplication en cis, dite duplication en tandem) ou d’une duplication totale du génome (duplication en trans) : un gène est dupliqué et forme deux copies filles, ou paralogues, qui sont à l’origine fonctionnellement redondantes. B. Recombinaison, à l’occasion d’une recombinaison non homologue ou d’une rétrotransposition : des parties codantes provenant de gènes différents fusionnent pour constituer un gène qui peut coder une protéine avec une nouvelle fonction. C. Transfert horizontal : transfert d’un gène d’une espèce à une autre ; ce mécanisme, très fréquent chez les bactéries, se produit également chez des eucaryotes, souvent en relation avec la phagocytose et le parasitisme qui rapprochent les génomes d’espèces très différentes. D. Création de novo : dans les génomes, il existe de très nombreuses séquences non codantes transcrites et qui contiennent un court cadre de lecture ouvert (ORF). Ces séquences, à l’origine non fonctionnelles, sont le plus souvent éliminées, car il n’y a pas de pression de sélection pour leur maintien. Elles sont aussi souvent associées à des séquences régulatrices qui contrôlent leur expression dans le temps et/ou l’espace. Si la protéine codée permet l’augmentation, aussi faible soit-elle, de la valeur sélective, le protogène peut être sélectionné et peut accumuler progressivement des modifications de la séquence codante et des séquences régulatrices jusqu’à former un nouveau gène qui aura une faible probabilité de disparaître au cours de l’évolution. Les séquences codantes sont figurées par des boîtes colorées, l’origine de transcription par une flèche brisée et les séquences régulatrices par des triangles de couleur (modifié d’après Carvunis et al. [7]). |
| Dans le texte | |
|
Figure 2. Phylostratigraphie des gènes de la souris. Les gènes présents dans le génome de la souris sont comparés aux gènes présents dans les génomes d’autres espèces afin d’identifier le moment de leur apparition. En s’appuyant sur la phylogénie des espèces, l’âge d’un gène est déterminé en identifiant l’ensemble des espèces qui partagent ce gène avec la souris. Par exemple, si un gène n’existe que chez la souris, il est apparu après la séparation de l’ancêtre du rat et de celui de la souris. Si un gène est partagé uniquement par la souris et les autres mammifères, il est apparu après la séparation des mammifères de toutes les autres espèces et avant le dernier ancêtre commun des mammifères. Si un gène est présent chez la souris et toutes les autres espèces, il était donc déjà présent chez LUCA, l’ancêtre commun de toutes les espèces. AC : ancêtre commun (modifié d’après Neme et al. [30]). |
| Dans le texte | |
|
Figure 3. Les deux voies de formation des nouveaux gènes. La plupart des nouveaux gènes qui apparaissent au sein des génomes sont formés par duplication, fusion ou transfert horizontal (boucle verte). Cependant, 5 à 12 % des gènes codant des protéines seraient formés de novo à partir d’ADN non codant. Ces nouveaux gènes apparaitraient progressivement ; des protogènes de plus en plus conformes aux propriétés des gènes constitueraient un gradient entre les deux états non-gène/gène. Cette création de gènes de novo serait contrebalancée par l’élimination continue de gènes (pseudogénisation) et de protogènes (modifié d’après Carvunis et al. [7]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.