")
")
| Issue |
Med Sci (Paris)
Volume 30, Number 5, Mai 2014
|
|
|---|---|---|
| Page(s) | 493 - 495 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20143005006 | |
| Published online | 13 juin 2014 | |
La sphingosine 1-phosphate comme biomarqueur de la maladie d’Alzheimer ?
Sphingosine 1-phosphate as a biomarker for Alzheimer’s disease?
1
Service d’anatomie pathologique, CHU Toulouse Rangueil, faculté de médecine, 1, avenue Jean Poulhès, 31059 Toulouse Cedex 9, France
2 Inserm UMR 1037, France
3 CNRS, institut de pharmacologie et de biologie structurale, Toulouse, France
4 Université de Toulouse, UPS, IPBS, Toulouse, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
La maladie d’Alzheimer est caractérisée par des lésions neuropathologiques : dépôts amyloïdes (Aβ), dégénérescences neurofibrillaires (DNF) et mort neuronale [1, 2]. Ces altérations se propagent progressivement et spécifiquement d’une région à l’autre du cerveau [3]. Au-delà de ces lésions classiques, l’implication éventuelle de nombreux facteurs dans les mécanismes cellulaires qui provoquent les signes cliniques de la maladie est à l’étude, ainsi que la recherche de nouvelles voies thérapeutiques [4].
Autrefois considérés comme de simples constituants membranaires, les sphingolipides sont aujourd’hui reconnus comme des molécules de signalisation à l’importance critique dans la détermination du devenir cellulaire, via la régulation de la balance mort/survie [5]. La dérégulation du rhéostat sphingolipidique a été décrite dans les cancers, mais qu’en est-il des implications physiopathologiques d’un tel système dans la maladie d’Alzheimer ? Les approches complémentaires de deux études récentes [6, 7] ont permis de préciser le rôle de ces lipides et ouvrent de nouvelles perspectives pour des stratégies diagnostiques ou thérapeutiques.
Le biostat sphingolipidique
Au cœur du biostat sphingolipidique se trouve la sphingosine 1-phosphate (S1P), issue de la métabolisation de la sphingosine, le composant principal de tous les sphingolipides (Figure 1). La S1P est produite en réponse à l’action des sphingosine kinases 1 et 2 (SphK1/SphK2) [8]. Son recyclage est assuré par des phosphatases alors que son élimination est prise en charge par la sphingosine 1-phosphate lyase (SPL). Au niveau intracellulaire, la S1P intervient dans des voies permettant par exemple d’activer le facteur NF-κB et de participer à la régulation épigénétique de l’ADN. Lorsqu’elle est sécrétée, la S1P exerce des effets autocrines et paracrines via cinq récepteurs couplés à la protéine G dont la répartition est spécifique des tissus [9].
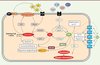 |
Figure 1. Représentation schématique du métabolisme sphingolipidique et de son rôle dans la maladie d’Alzheimer. Le céramide produit de novo ou par hydrolyse de la sphingomyéline stabiliserait BACE-1 (beta-site amyloid precursor protein cleaving enzyme 1) dans la maladie d’Alzheimer. La surproduction d’A β aussi induite via l’activation du récepteur p75NTR (p75 neurotrophin receptor) provoque une augmentation de la production de céramide. Ce cercle vicieux serait à l’origine d’une amplification de la signalisation apoptotique induite par le céramide et le peptide Aβ. Le céramide est également métabolisé en sphingosine puis en S1P grâce à l’intervention des sphingosine kinases 1 et 2. Elles peuvent être activées par différents facteurs de croissance. La S1P sécrétée exerce une action paracrine et autocrine via cinq récepteurs couplés à protéine G ; l’effet produit dépend du profil d’expression des récepteurs à la surface de la cellule. La S1P intracellulaire mobilise les réserves calciques, active TRAF2 (TNF receptor-associated factor 2) et NF-κB et régule l’activation de gènes via l’inhibition des histones déacétylases 1 et 2 (HDAC 1/2). La dégradation définitive de S1P assurée par la SPL génère de l’hexadécénal et de l’éthanolamine phosphate dont les effets ne sont pas encore totalement élucidés. Ces produits de dégradation pourraient jouer un rôle important dans les syndromes neuropathologiques. SMS : sphingomyéline synthase ; Smase : sphingomyélinase ; Cerase : céramidase ; CerS : céramide synthase ; Sph : sphingosine ; SphK1/2 : sphingosine kinase 1/2 ; S1Pase : sphingosine 1-phosphate phosphatase ; SPL : sphingosine 1-phosphate lyase. |
Dans un tissu normal, la concentration en S1P est maintenue à un faible niveau via un équilibre délicat entre sa synthèse et sa dégradation. Dans le cancer, le niveau de S1P augmente, ce qui est notamment associé à la prolifération cellulaire et à la résistance aux traitements. Une étude récente a montré que, dans le cancer de la prostate, la dérégulation du niveau de S1P était causée non seulement par une augmentation de l’activité et de l’expression de la SphK1 (l’isoforme la plus étudiée), mais aussi par une perte d’expression et d’activité enzymatique de la SPL [10]. La manipulation de l’expression de ces deux enzymes dans des modèles cellulaires a permis de moduler la réponse à la chimiothérapie et à la radiothérapie, et de diminuer la prolifération cellulaire via la régulation du niveau de S1P, confirmant le rôle fondamental de ce sphingolipide dans la tumorigenèse. Si une augmentation anormale du niveau de S1P peut induire une prolifération cellulaire, sa diminution pourrait-elle être à l’origine d’un comportement opposé, proapoptotique, comme cela est observé dans les maladies neurodégénératives ? C’est cette hypothèse qu’explorent un nombre croissant de travaux analysant la dérégulation du biostat sphingolipidique dans la maladie d’Alzheimer.
Lipides et maladie d’Alzheimer
Apolipoprotéine E et céramide
L’étude des lipides dans la maladie d’Alzheimer n’est pas un sujet nouveau, si l’on se réfère par exemple à l’apolipoprotéine E (ApoE) qui est un facteur de prédisposition bien connu [11]. En revanche, les premiers travaux sur les sphingolipides sont relativement récents et surtout focalisés sur le métabolisme du céramide, un précurseur de la S1P. Si la littérature s’accorde sur l’effet proapoptotique du céramide, son niveau d’expression dans la maladie d’Alzheimer est plus controversé [12]. Récemment, Couttas et ses collaborateurs ont montré que l’expression d’une seule des différentes espèces de céramide (C16) était modestement augmentée dans la région temporale du cerveau de patients atteints de maladie d’Alzheimer, en fonction du score de Braak [6]. Cette classification permet d’évaluer le degré de progression de la maladie via l’étude de l’extension des dégénérescences neurofibrillaires dans les différents territoires cérébraux [3]. Le céramide n’est donc pas le seul acteur de la mort neuronale observée dans la maladie d’Alzheimer, comme le suggèrent les données récentes sur la S1P.
S1P et maladie d’Alzheimer, un problème de synthèse ?
Une équipe australienne vient d’établir que le niveau de S1P était inversement corrélé au stade de Braak chez les patients atteints de la maladie d’Alzheimer [6]. Cette variation de la concentration en S1P dans certaines régions du cerveau (lobe temporal et hippocampe) était directement liée à une diminution de l’activité des enzymes SphK1 et SphK2 [6]. Parallèlement, nos travaux ont montré une diminution de l’activité de la SphK1 dans des cellules exposées au peptide Aβ (25-35) [13].
Nos travaux plus récents sur des tissus humains ont montré que l’expression de la SphK1 et d’un des récepteurs impliqués dans son activation (IGF-1R) était nettement diminuée chez les patients atteints de la maladie d’Alzheimer comparativement à des sujets non déments du même âge [7]. Une étude immunohistochimique détaillée des tissus cérébraux a également mis en évidence une corrélation inverse entre l’expression de la SphK1 et la densité des dépôts amyloïdes dans le cortex entorhinal (région parahippocampique impliquée dans la formation de la mémoire) [7]. Ces données originales indiquent que la diminution du taux de S1P dans la maladie d’Alzheimer résulte d’une dérégulation des enzymes responsables de sa synthèse, directement corrélée au développement des lésions principales de cette maladie (dégénérescences neurofibrillaires et dépôts amyloïdes).
S1P et maladie d’Alzheimer, un problème de dégradation ?
Les voies de dégradation de la S1P ont été également explorées. Couttas et al. ont montré que l’activité des phosphatases responsables du recyclage de la S1P était corrélée au stade de Braak dans le lobe temporal. Nos travaux ont montré que la SPL était également dérégulée dans la maladie d’Alzheimer, puisque son expression était significativement augmentée dans le cerveau des malades et que, dans le cortex entorhinal, elle était positivement corrélée à la densité des dépôts amyloïdes [7]. La dérégulation de la S1P dans la maladie d’Alzheimer ne peut donc pas être attribuée seulement à un défaut de synthèse, mais elle s’explique également par une dégradation anormalement élevée. Si l’on se réfère à ces travaux, et à ceux de Brizuela et al. dans le cancer [10], la SphK1 et la SPL apparaissent comme deux acteurs principaux de cette dérégulation ; toutes deux sont modulées dans la maladie d’Alzheimer de manière diamétralement opposée à ce qui est observé dans le cancer. En effet, la dérégulation de la balance sphingolipidique conduit dans le cancer à une augmentation de la S1P et à une prolifération cellulaire, et, dans la maladie d’Alzheimer, à une diminution de la S1P qui participe à la dégénérescence des neurones.
Nous avons exploré la dérégulation d’un troisième protagoniste, le S1P1, un récepteur majeur de S1P, présent dans les neurones. L’expression de ce récepteur est significativement diminuée dans le cerveau des patients atteints de la maladie d’Alzheimer, ce qui traduit un défaut de signalisation en réponse à la S1P sécrétée. Or, récemment, l’administration d’un agoniste des récepteurs S1P1,3,4,5 (FTY720, Fingolimod, GilyenaTM) a entraîné une certaine correction des déficits mnésiques observés dans un modèle murin de la maladie d’Alzheimer [14]. Une étude électrophysiologique dans un modèle animal a également montré que le récepteur S1P3 et le niveau de S1P dans la région CA3 de l’hippocampe étaient des déterminants majeurs du mécanisme de potentialisation à long terme, qui est aujourd’hui considéré comme une des bases moléculaires de la formation de la mémoire [15]. Ces données soulignent l’importance des voies de signalisation activées par les sphingolipides, non seulement pour la survie des cellules, mais également pour assurer le bon fonctionnement des réseaux neuronaux.
Conclusion
Les données obtenues dans ces deux études indiquent que la dérégulation du biostat sphingolipidique dans la maladie d’Alzheimer obéit à un schéma de propagation spatio-temporelle parallèle à celui suivi par les lésions typiques de cette maladie [6, 7]. D’autre part, ces travaux montrent que la dérégulation du métabolisme de la S1P repose à la fois sur un défaut de synthèse et sur une augmentation de sa dégradation. Enfin, ils offrent de nouvelles perspectives diagnostiques (mesure du taux de S1P dans le liquide céphalo-rachidien) et pourquoi pas thérapeutiques, s’il est possible d’agir simultanément sur les acteurs de la synthèse et de la dégradation de la S1P, mais aussi sur ses récepteurs.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol 2009 ; 118 : 5–36. [CrossRef] [PubMed] [Google Scholar]
- Campion D, Hannequin D. La duplication du gène APP cause de maladie d’Alzheimer associée à une importante angiopathie amyloïde. Med Sci (Paris) 2006 ; 22 : 468–469. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006 ; 112 : 389–404. [CrossRef] [PubMed] [Google Scholar]
- Helmer C, Pasquier F, Dartigues JF. Épidémiologie de la maladie d’Alzheimer et des syndromes apparentés. Med Sci (Paris) 2006 ; 22 : 288–296. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Cuvillier O, Pirianov G, Kleuser B, et al. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996 ; 381 : 800–803. [CrossRef] [PubMed] [Google Scholar]
- Couttas TA, Kain N, Daniels B, et al. Loss of the neuroprotective factor Sphingosine 1-phosphate early in Alzheimer’s disease pathogenesis. Acta Neuropathol Commun, 2014 ; 2 : 9. [CrossRef] [PubMed] [Google Scholar]
- Ceccom J, Loukh N, Lauwers-Cances V, et al. Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta Neuropathol Commun, 2014 ; 2 : 12. [CrossRef] [PubMed] [Google Scholar]
- Pitson SM. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci 2011 ; 36 : 97–107. [CrossRef] [PubMed] [Google Scholar]
- Cuvillier O. Les récepteurs de la sphingosine 1-phosphate. De la biologie à la physiopathologie Med Sci (Paris) 2012 ; 28 : 951–957. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Brizuela L, Ader I, Mazerolles C, et al. First evidence of sphingosine 1-phosphate lyase protein expression and activity downregulation in human neoplasm: implication for resistance to therapeutics in prostate cancer. Mol Cancer Ther 2012 ; 11 : 1841–1851. [CrossRef] [PubMed] [Google Scholar]
- Gosselet F. Apolipoprotéine E et intégrité de la barrière hémato-encéphalique. Med Sci (Paris) 2012 ; 28 : 920–923. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Mielke MM, Lyketsos CG. Alterations of the sphingolipid pathway in Alzheimer’s disease: new biomarkers and treatment targets? Neuromolecular Med 2010 ; 12 : 331–340. [CrossRef] [PubMed] [Google Scholar]
- Gomez-Brouchet A, Pchejetski D, Brizuela L, et al. Critical role for sphingosine kinase-1 in regulating survival of neuroblastoma cells exposed to amyloid-beta peptide. Mol Pharmacol 2007 ; 72 : 341–349. [CrossRef] [PubMed] [Google Scholar]
- Hemmati F, Dargahi L, Nasoohi S, et al. Neurorestorative effect of FTY720 in a rat model of Alzheimer’s disease: Comparison with Memantine. Behav Brain Res 2013 ; 252C : 415–421. [CrossRef] [Google Scholar]
- Kanno T, Nishizaki T, Proia RL, et al. Regulation of synaptic strength by sphingosine 1-phosphate in the hippocampus. Neuroscience 2010 ; 171 : 973–980. [CrossRef] [PubMed] [Google Scholar]
© 2014 médecine/sciences – Inserm
Liste des figures
|
Figure 1. Représentation schématique du métabolisme sphingolipidique et de son rôle dans la maladie d’Alzheimer. Le céramide produit de novo ou par hydrolyse de la sphingomyéline stabiliserait BACE-1 (beta-site amyloid precursor protein cleaving enzyme 1) dans la maladie d’Alzheimer. La surproduction d’A β aussi induite via l’activation du récepteur p75NTR (p75 neurotrophin receptor) provoque une augmentation de la production de céramide. Ce cercle vicieux serait à l’origine d’une amplification de la signalisation apoptotique induite par le céramide et le peptide Aβ. Le céramide est également métabolisé en sphingosine puis en S1P grâce à l’intervention des sphingosine kinases 1 et 2. Elles peuvent être activées par différents facteurs de croissance. La S1P sécrétée exerce une action paracrine et autocrine via cinq récepteurs couplés à protéine G ; l’effet produit dépend du profil d’expression des récepteurs à la surface de la cellule. La S1P intracellulaire mobilise les réserves calciques, active TRAF2 (TNF receptor-associated factor 2) et NF-κB et régule l’activation de gènes via l’inhibition des histones déacétylases 1 et 2 (HDAC 1/2). La dégradation définitive de S1P assurée par la SPL génère de l’hexadécénal et de l’éthanolamine phosphate dont les effets ne sont pas encore totalement élucidés. Ces produits de dégradation pourraient jouer un rôle important dans les syndromes neuropathologiques. SMS : sphingomyéline synthase ; Smase : sphingomyélinase ; Cerase : céramidase ; CerS : céramide synthase ; Sph : sphingosine ; SphK1/2 : sphingosine kinase 1/2 ; S1Pase : sphingosine 1-phosphate phosphatase ; SPL : sphingosine 1-phosphate lyase. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.