")
")
| Issue |
Med Sci (Paris)
Volume 30, Number 4, Avril 2014
|
|
|---|---|---|
| Page(s) | 445 - 451 | |
| Section | Microenvironnements tumoraux : conflictuels et complémentaires | |
| DOI | https://doi.org/10.1051/medsci/20143004021 | |
| Published online | 5 mai 2014 | |
Le microenvironnement tumoral et la résistance thérapeutique
Tumor microenvironment and therapeutic resistance process
1
Département de pédiatrie, de biochimie et biologie moléculaire
2
Keck school of medicine, university of Southern California, États-Unis
3
Children’s hospital Los Angeles, 4650 Sunset Boulevard CA
90027
Los Angeles, California, États-Unis
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Au cours de la dernière décennie, il est devenu évident que le cancer n’est pas seulement une maladie causée par des gènes anormaux. Ainsi, le tissu avoisinant les cellules cancéreuses joue également un rôle important. De nombreuses interactions entre les cellules cancéreuses et le stroma tumoral, qui consiste en une matrice extracellulaire (MEC) et des cellules bénignes (appelées cellules stromales), favorisent la survie cellulaire et la résistance à la thérapie. Ces mécanismes sont maintenant connus et dépendent, soit d’un contact entre les cellules cancéreuses et la MEC ou les cellules stromales, soit de facteurs solubles ou de microvésicules. La moelle osseuse joue un rôle particulièrement important dans la résistance aux thérapies en tant que sanctuaire privilégié protégeant les cellules cancéreuses des effets toxiques de la chimiothérapie, mais aussi en tant que source de nombreuses cellules stromales qui colonisent la tumeur primaire et forment la niche prémétastatique. Cette meilleure connaissance des mécanismes par lesquels le microenvironnement tumoral favorise la résistance thérapeutique a conduit à des essais cliniques avec des agents qui ont pour fonction d’interférer avec les interactions entre cellules cancéreuses et stroma. Cette nouvelle voie de recherche clinique est particulièrement prometteuse.
Abstract
Over the last decade, it has become clear that cancer is not just a disease of the genes, and that the tumor microenvironment (TME) plays an important role in cancer progression. Interactions between tumor cells and the TME, made of the extracellular matrix (ECM) and of non-transformed cells (designated here as stromal cells), promote cancer cell survival and drug resistance. Many of the mechanisms involved are known and are either contact-dependent or contact-independent. Contact between tumor cells and the ECM or stromal cells as well as the production of soluble factors and microvesicles all contribute. The bone marrow plays a special role in environment-mediated drug resistance as it is not only a sanctuary protecting tumor cells from cytotoxic drugs, but also a source of many stromal cells that colonize primary tumors and contribute to the pre-metastatic niche. As our understanding of the mechanisms by which the tumor microenvironment promotes therapeutic resistance progresses, clinical trials testing agents that disrupt the interaction between tumor cells and the stroma have been initiated. This new avenue of therapy is promising.
Cet article fait partie du numéro thématique publié par médecine/sciences en avril 2014 et intitulé « Microenvironnement tumoral ».
© 2014 médecine/sciences – Inserm
Une des principales raisons de notre incapacité à éradiquer le cancer vient de l’énorme aptitude des cellules cancéreuses à muter, en raison de son instabilité. Ceci explique que ces cellules deviennent génétiquement résistantes aux agents auxquels elles ont été exposées. Cette capacité d’acquérir une résistance au traitement réside dans l’expression de gènes impliqués dans le transport cellulaire des agents chimiothérapeutiques et dans l’apparition de mutations qui rendent une protéine résistante à l’agent thérapeutique avec lequel elle interagit. C’est le cas de la résistance à l’imatinib1 des cellules portant la protéine de fusion BCR/ABL, liée au développement de voies de signalisation qui prennent le relais de la voie affectée par le traitement [45] (→). Dans tous ces cas, la raison première de la résistance thérapeutique est la cellule cancéreuse elle-même.
(→) Voir la Synthèse de J.C. Chomel et al., page 452 de ce numéro
Au cours de ces dernières années, il est devenu évident que la résistance thérapeutique n’est pas seulement le résultat de changements génétiques qui se développent dans les cellules cancéreuses, mais qu’elle est également due à des changements dans le microenvironnement tumoral. Cette notion dite de « résistance relayée par le microenvironnement tumoral » est fondée sur l’existence d’interactions entre la cellule cancéreuse et, soit la matrice extracellulaire (MEC), soit les cellules qui ne sont pas atteintes par le processus tumoral (désignées ici sous le nom de cellules stromales) [46, 47] (→). Nous récapitulons dans cette revue les différents mécanismes par lesquels le microenvironnement tumoral influence la réponse au traitement (Figure 1). Le rôle particulier de la moelle osseuse en tant que sanctuaire protégeant les cellules cancéreuses des effets des traitements anticancéreux sera ensuite discuté. En dernier lieu, les possibilités d’intervention thérapeutiques seront présentées.
(→) Voir les Synthèses de W.H. Fridman et C. Sautès-Fridman, et de V. Provot, pages 359 et 366 de ce numéro
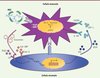 |
Figure 1. Interactions entre cellules tumorales et cellules mésenchymateuses du stroma. Ces interactions et les voies de signalisation décrites favorisent la survie des cellules tumorales et la résistance aux thérapies anticancéreuses. EP : prostaglandin E2 receptor. BIM : membre de la famille Bcl2 proapoptotique ; FLIP : FLICE-like inhibitory protein (antiapoptotique) ; VEGF : vascular endothelial growth factor ; FGF : fibroblast growth factor ; PGE2 : prostaglandine E2. |
Mécanismes responsables de la résistance thérapeutique
On reconnaît trois mécanismes de communication entre les cellules cancéreuses et le microenvironnement, responsables de la résistance thérapeutique.
Adhérence à la matrice extracellulaire
Fridman et al. démontrèrent les premiers que l’attachement d’une cellule tumorale à la MEC stimule la prolifération cellulaire et la résistance thérapeutique. Ils montrèrent en 1990 que des cellules de cancer du poumon deviennent résistantes à des agents chimiothérapeutiques comme le cisplatinum, la doxorubicine ou l’étoposide quand elles sont cultivées sur de la laminine. Ils montrèrent également que cette résistance disparaissait en présence d’un peptide (YIGSR) inhibiteur de l’adhérence [1]. Quelques années plus tard, Damiano et al. firent une observation similaire avec des cellules de myélome : le blocage de l’interaction de l’intégrine α4β1 (VLA 4) ou α5β1 (VLA5) avec la fibronectine ou le collagène augmentait la sensibilité des cellules cancéreuses à la doxorubicine ou au melphalan [2]. Cet effet protecteur de la MEC ne semble donc pas dépendant de la nature de la protéine extracellulaire. De plus, ce n’est pas seulement la résistance à la chimiothérapie qui est affectée, mais aussi la résistance à la radiothérapie, comme l’illustre l’expérience de Park et al. [3]. Cet effet n’est pas seulement démontré in vitro mais aussi in vivo. Hsieh et al. ont administré un inhibiteur bloquant l’intégrine α4 à des souris transplantées avec des cellules leucémiques humaines exprimant BCR /ABL. Ces cellules non seulement devenaient plus sensibles au nilotinib, un inhibiteur de BCR/ABL, mais elles répondaient mieux à la chimiothérapie [4]. Les mécanismes par lesquels les intégrines protègent les cellules cancéreuses sont connus. L’adhérence relayée par l’intégrine β1 induit une résistance des cellules leucémiques et myélomateuses à l’apoptose extrinsèque causée par Fas (CD95). Ceci est associé à une redistribution de la protéine anti-apoptotique FLIP (FADD-like apoptosis regulator), à une augmentation de la dégradation des protéines proapoptotiques comme BIM (Bcl2 interacting mediator of cell death protein), à une augmentation de l’expression de la protéine p27 et à une inhibition des kinases impliquées dans le cycle de prolifération cellulaire. Il s’ensuit un arrêt de prolifération [5, 6]. Ces observations illustrent plusieurs points importants : (1) l’adhérence cellulaire à la MEC a un effet double : elle favorise la survie et bloque la prolifération ; (2) ces effets sont relayés par des modifications post traductionnelles des protéines, et non pas au niveau de leur synthèse, permettant une réponse rapide et réversible ; (3) bien que ces effets aient été principalement décrits dans le contexte d’une résistance à la chimiothérapie, ils sont également impliqués dans la résistance aux molécules qui interfèrent spécifiquement avec les kinases de signalisation cellulaire.
Contact entre les cellules cancéreuses et stromales
Un second mécanisme de résistance implique le contact direct entre les cellules cancéreuses et les cellules du stroma. L’adhérence intercellulaire, par l’intermédiaire de protéines comme VCAM (vascular cell adhesion molecule) ou l’intégrine β1, joue un rôle important dans la résistance thérapeutique [7, 8]. Par exemple, l’adhérence de cellules tumorales myélomateuses aux cellules stromales de la moelle osseuse par l’intermédiaire de l’intégrine β1 stimule leur production d’interleukine-6 (IL-6) qui, à son tour, protège les cellules cancéreuses [9]. Ces cellules cancéreuses, mises en contact avec des hépatocytes, expriment de nouveau la E-cadhérine, favorisant leur chimiorésistance [10]. Le contact entre les cellules leucémiques et la niche de la moelle osseuse favorise leur résistance aux inhibiteurs de kinases, comme le sorafenib et le SU5614 [11].
Facteurs solubles
De nombreuses expériences de coculture de cellules stromales et cancéreuses ont démontré qu’en présence de ces dernières, la production de facteurs solubles, comme en particulier l’IL-6 et la chimiokine stromal-derived factor (SDF-1 ou CXCL12) par les cellule stromales augmentait.
L’action paracrine de l’IL-6
Notre laboratoire a été parmi les premiers à démontrer que les cellules des métastases osseuses de neuroblastome2 stimulent l’expression d’IL-6 par les cellules mésenchymateuses de la moelle osseuse [12]. De plus, l’IL-6 stimule l’activité des ostéoclastes et l’expression dans les cellules cancéreuses de plusieurs protéines de survie, comme la survivine, Bcl-XL et Mcl-1 (myeloid cell leukemia protein-1), qui les rendent résistantes à des agents tels que l’étoposide ou le melphalan [13]. L’IL-6 augmente également l’expression de FLIP [14]. Ce mécanisme met en jeu l’activation (phosphorylation) du facteur STAT3 (signal transducer and activator of transcription 3) (pSTAT3) en réponse à la fixation de l’IL-6 à son récepteur IL-6R (composé de deux protéines, gp80 et gp130). pSTAT3 migre ensuite vers le noyau cellulaire où il agit comme facteur de transcription [15]. Il faut souligner que, dans le cas du neuroblastome, il s’agit bien d’un mécanisme paracrine puisque les cellules du neuroblastome ne produisent pas d’IL-6 (qui n’est synthétisée que par les cellules stromales). Parmi ces cellules, on compte non seulement les cellules mésenchymateuses de la moelle, mais aussi les monocytes et les macrophages qui, outre l’IL-6, produisent le récepteur soluble de cette cytokine, qui agit comme agoniste et potentialise l’effet de l’IL-6. La production d’IL-6 par les cellules stromales de moelle joue un rôle similaire dans le myélome multiple. Mais, dans le cas du myélome, la stimulation des cellules tumorales par l’IL-6 des cellules stromales est le résultat d’un contact cellulaire direct, et non pas d’une interaction avec des protéines solubles.
Le rôle de SDF-1
SDF-1 (CXCL12) est un autre facteur soluble important [16]. Cette chimiokine est sécrétée par de nombreuses cellules mésenchymateuses, en particulier par les ostéoblastes. Elle joue un rôle important dans la maintenance du pool de cellules souches hématopoïétiques qui expriment le récepteur du SDF-1 (CXCR4 ou C-X-C chemokine receptor type 4) [17]. Le récepteur CXCR4 est également exprimé par de nombreuses cellules tumorales qui peuvent donc entrer en compétition avec les cellules souches hématopoïétiques pour la niche ostéoblastique [18]. L’interaction SDF-1/CXCR4 dans les cellules tumorales permet une localisation de ces dernières dans la moelle osseuse, souvent sous forme de cellules dormantes, qui leur permet ainsi d’échapper aux effets toxiques des agents chimiothérapeutiques [19]. À nouveau, il faut souligner que les facteurs solubles produits par les cellules stromales ne protègent pas seulement des effets de la chimiothérapie, mais aussi des effets d’agents qui bloquent des voies spécifiques de signalisation contrôlant la survie cellulaire, tels que SDF-1 qui protège les cellules cancéreuses des effets apoptotiques d’un inhibiteur de mTOR (mammalian target of rapamycin) [20]. Le fait que SDF-1 augmente également l’expression de l’intégrine β1 et l’adhésion des cellules cancéreuses à la MEC (augmentant ainsi la résistance thérapeutique [21]), illustre bien l’interaction qui existe entre les facteurs solubles et les mécanismes dépendant de l’adhésion. La production d’autres facteurs solubles comme le VEGF (vascular endothelial growth factor), le FGF (fibroblast growth factor), le MCP-1 (monocyte chemotactic protein-1) et le TGF-β (transforming growth factor-β) par les cellules stromales en présence de cellules tumorales a aussi été rapportée. Ces facteurs semblent protéger les cellules de l’apoptose, mais les mécanismes impliqués ne sont pas entièrement connus. Par exemple, en stimulant la transformation épithélio-mésenchymateuse, le TGF-β stimule un phénotype cancéreux résistant au traitement [22]. Le VEGF, de son côté, diminue la diffusion des agents thérapeutiques dans le tissu interstitiel et leur accès aux cellules tumorales, en favorisant la formation d’un réseau vasculaire désorganisé [23]. Le VEGF et le FGF peuvent aussi avoir une action indirecte en stimulant la production d’IL-6 par les cellules stromales [24].
Éducation des cellules stromales par les cellules cancéreuses
En 2003, Nefedova et al. ont rapporté l’observation suivante : le milieu de culture de cellules mésenchymateuses de moelle osseuse induit, dans les cellules de myélome, une résistance à certaines molécules thérapeutiques, à la condition que les cellules mésenchymateuses soient d’abord cultivées en présence de cellules tumorales [25]. Cette observation démontre que les cellules stromales sont « éduquées » par les cellules cancéreuses, et elle soulève la question des mécanismes par lesquels les cellules cancéreuses influencent les cellules stromales. Ici, de nouveau, les facteurs solubles jouent un rôle important. L’effet final est en général la production d’une réaction inflammatoire de type T helper (Th2), que l’on observe dans les maladies parasitaires et qui se caractérise par une élévation de la production d’IL-4, d’IL-10 et d’IL-13, une diminution de la production d’IL-12 et des changement métaboliques comme une augmentation d’arginase. Cette réaction inflammatoire favorise le développement tumoral et conduit à une tolérance immunitaire. Les facteurs produits par les cellules tumorales qui influencent les cellules stromales sont multiples, mais certains méritent d’être plus particulièrement mentionnés. Notre laboratoire a démontré que la protéine Gal-3BP, liée à la galectine-3 (Gal-3) et produite par les cellules de neuroblastome dans le milieu extracellulaire, joue un rôle important. Gal-3BP interagit avec la Gal-3 qui est présente dans les cellules mésenchymateuses et le complexe active la voie de signalisation Ras/MEK (MAP kinase kinase)/ERK (extracellular signal regulated kinase) responsable de l’activation du promoteur de l’IL-6 [26]. Les prostaglandines, comme PGE2, jouent également un rôle. À l’état basal, PGE2 est produite en faible concentration par les cellules de neuroblastome mais, en présence d’IL-6 qui active COX2 (cyclooxygenase 2), la production de PGE2 est fortement augmentée. À son tour, PGE2 stimule l’expression de l’IL-6 dans les cellules mésenchymateuses, créant donc une réaction d’amplification dans le microenvironnement tumoral [13]. VEGF (vascular endothelial growth factor) joue également un rôle en stimulant la production du SDF-1 par les cellules stromales [16], ce qui induit une résistance au traitement dans les cellules cancéreuses exprimant le récepteur CXCR4.
Tout récemment, l’attention s’est portée sur les microvésicules et, en particulier, les exosomes, produits par les cellules tumorales [27, 48] (→). Les exosomes, qui ont un diamètre de 10 à 100 microns, dérivent d’endosomes intracellulaires. Ces vésicules contiennent de nombreuses protéines, en particulier des protéines de stress comme Hsp90 (heat shock protein of 90-kDa) et Hsp70, mais aussi d’autres protéines intracellulaires et membranaires, des facteurs de transcription, des enzymes, des facteurs métaboliques, ainsi que des microARN. Ils ont d’abord été identifiés par leur rôle critique dans la réaction du système immunitaire et comme source d’antigènes [28]. Plus récemment, leur rôle a été démontré dans la communication entre cellules tumorales et cellules stromales et, en particulier, dans l’établissement de la niche prémétastatique [29, 30, 47] (→→). Le fait que ces exosomes contiennent des microARN laisse envisager un nouveau mécanisme de communication faisant intervenir les microARN dans le contrôle de la régulation des cytokines et chimiokines qui affectent la résistance thérapeutique. Par exemple, Fabbri et al. ont récemment démontré que les exosomes produits par des cellules de cancer du poumon stimulent la production d’IL-6 par des cellules myéloïdes, selon un effet dépendant du Toll-like récepteur TLR8 [31].
(→) Voir la Synthèse de F.A. Fellouse, page 405 de ce numéro
(→→) Voir la Synthèse de S. Provot, page 366 de ce numéro
La moelle osseuse, sanctuaire et contrôleur de l’environnement tumoral et métastatique
La moelle osseuse joue un rôle tout particulier dans le microenvironnement tumoral (Figure 2). Tout d’abord, elle est riche en protéines extracellulaires, comme le collagène et la fibronectine, et elle est composée de cellules souches mésenchymateuses et hématopoïétiques, et de leurs descendants [32]. La moelle osseuse forme une niche pour les cellules cancéreuses, qui leur permet de survivre en état de latence et d’être réactivées plus tard sous forme de colonies prolifératives et résistantes au traitement [49] (→). La moelle est aussi riche en facteurs solubles comme l’IL-6, le SDF-1, le VEGF, le FGF et le TGF-β, qui tous contribuent à la survie des cellules cancéreuses et à leur résistance à la thérapeutique [19]. En favorisant la survie de cellules cancéreuses en présence d’agents thérapeutiques, l’environnement de la moelle osseuse permet à ces cellules ayant acquis une résistance extrinsèque et réversible, de développer – à la suite de mutations critiques – une résistance intrinsèque devenue alors irréversible. Ce concept est au cœur de la théorie adaptative du cancer. Dans ce modèle, les propriétés intrinsèques liées aux mutations (sélectives) ne sont pas en elles-mêmes suffisantes pour expliquer l’apparition d’un cancer ou d’une cellule cancéreuse résistante à une chimiothérapie particulière. Il faut aussi l’intervention d’un milieu favorable à la sélection des cellules résistantes. Cette théorie du « sol et de la semence » (soil and seed), proposée par Paget en 1889 [33], permet de comprendre la grande variabilité que l’on observe dans l’apparition de cellules résistantes dans la moelle ou dans le sang périphérique de patients atteints de cancer [34]. De plus, il faut souligner que les cellules stromales d’une tumeur ont pour origine soit les tissus avoisinants (cellules résidentes), soit la moelle osseuse. Cette dernière agit donc comme contrôleur à distance de la tumeur primaire et des niches prémétastatiques [35]. De nombreuses cellules souches de la moelle, comme les cellules souches mésenchymateuses et myélomonocytaires, et les précurseurs des cellules endothéliales, sont recrutées par la tumeur primaire qu’elles colonisent. Elles contribuent à la présence de fibroblastes tumoraux, de macrophages tumoraux, et de cellules endothéliales et inflammatoires [36] qui créent un milieu favorable à la croissance des cellules cancéreuses, à leur survie et à leur résistance thérapeutique. De nombreuses expériences récentes ont démontré que la présence de cellules mésenchymateuses et de macrophages dans une tumeur favorise la résistance à la chimiothérapie et à la radiothérapie [37, 38]. Certaines cellules de la moelle osseuse, comme les cellules hématopoïétiques exprimant le récepteur VEGFR1, ont également la capacité de coloniser les tissus périphériques et de les rendre davantage sensibles au recrutement de cellules tumorales [39, 50] (→→). La déposition de fibronectine est, entre autres, un élément important, ce qui suggère (compte tenu de son rôle dans l’induction de la résistance thérapeutique expliqué plus haut) que ces niches offrent aux cellules circulantes tumorales l’opportunité d’être protégées de la chimiothérapie.
(→) Voir la Nouvelle de J. Bensimon, m/s n° 12, décembre 2013, page 1069
(→→) Voir la Synthèse de S. Hubert et J.P. Abastado, page 378 de ce numéro
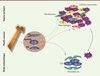 |
Figure 2. Contrôle de l’environnement tumoral et métastatique par la moelle osseuse. CSH : cellule souche hématopoïétique ; CSM : cellule souche mésenchymateuse ; PCE : précurseur endothélial ; CE : cellule endothéliale. |
Implications thérapeutiques
La question importante qui reste à élucider est celle de l’application des connaissances acquises dans le domaine du microenvironnement tumoral à la découverte de nouvelles approches pour le traitement anticancéreux. Sur le plan théorique, il est logique de penser que modifier « le sol » pour prévenir la croissance de la tumeur est une approche prometteuse (Tableau I). C’est ce qui fut démontré avec les essais cliniques de l’Avastin, cet anticorps anti-VEGFA qui a été approuvé aux États-Unis en 2004 [40]. En même temps, notre expérience avec les inhibiteurs nous a aussi appris qu’attaquer seulement « le sol » est insuffisant pour contrôler la croissance de la « semence », et que la résistance peut apparaître tant chez l’un que chez l’autre. Par exemple, lorsqu’elles sont exposées à l’Avastin, les cellules endothéliales deviennent plus sensibles à d’autres facteurs angiogéniques, comme le PlGF (placental growth factor), le PDGF (plateled-derived growth factor) et l’EGF (epidermal growth factor), en augmentant l’expression à leur surface des récepteurs pour ces facteurs de croissance [41]. La stratégie idéale si l’on veut utiliser des agents dont l’action est de bloquer un ou plusieurs mécanismes favorisant la résistance thérapeutique est probablement de les combiner avec une chimiothérapie ou une thérapie ciblée, afin de prévenir l’émergence de cellules cancéreuses résistantes [29]. Cette approche est actuellement testée avec des inhibiteurs d’intégrine comme le Cilengitide (inhibiteur cyclique de l’intégrine αv), qui est actuellement en essai clinique de phase II, en combinaison avec la radiothérapie et le temozolomide chez des patients atteints de glioblastome multiforme [42]. Un autre agent prometteur, le Plerixafor (AMD 3100, un antagoniste des récepteurs CXCR4), est employé de manière routinière en association avec le G-CSF (granulocyte colony-stimulating factor) pour mobiliser les cellules hématopoïétiques souches de la moelle osseuse vers le sang périphérique. Dans des modèles précliniques de souris auxquelles on a injecté des cellules leucémiques, cet agent augmente la mobilisation des cellules leucémiques de la moelle vers le sang périphérique et, en les délogeant de l’environnement protecteur de la moelle, stimule leur prolifération et leur sensibilité à la chimiothérapie et à l’imatinib [43, 44]. Contrôler l’inflammation soit par des anti-inflammatoires, soit par des agents qui inhibent le recrutement de macrophages ou qui préviennent leur polarisation vers un phénotype Th2, est une autre voie prometteuse.
Agents thérapeutiques agissant sur le microenvironnement tumoral approuvés ou en cours d’étude clinique.
Conclusion
Des progrès importants ont été accomplis aux cours des dernières années dans les connaissances du microenvironnement tumoral et de son rôle dans la progression du cancer. Ces connaissances ont révélé sa fonction importante dans le développement de la résistance thérapeutique et l’établissement de cellules cancéreuses dormantes. Les mécanismes de communication cellules cancéreuses/matrice extracellulaire/cellules stromales étant mieux connus, il devient possible d’intervenir d’un point de vue thérapeutique. La performance des essais cliniques combinant un ou plusieurs inhibiteurs de la résistance environnementale au traitement nous ouvre une voie prometteuse dans nos espoirs pour lutter contre le cancer.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Les travaux des auteurs décrits dans ce manuscrit ont été soutenus financièrement en partie par des fonds de recherche du National institutes of health U54 CA163117 et P01 CA081403 à Y. A. DeClerck. Les auteurs souhaitent remercier le docteur M. Torres pour son assistance et ses conseils dans la rédaction de cet article.
La mutation BCR/ABL est caractéristique des cellules de leucémie myéloïde chronique ; cette translocation réciproque et équilibrée entraîne la formation d’une protéine de fusion douée d’une activité tyrosinekinase délètère qui peut être contrecarrée par des inhibiteurs spécifiques, dont l’imatinib (ou Glivec).
Ces tumeurs se développent à partir des cellules originaires des tissus qui donnent naissance au système nerveux sympathique. Elles peuvent, de ce fait, être retrouvées sur toute la longueur de la colonne vertébrale et dans la partie interne de la glande surrénale. Rencontrées le plus souvent chez l’enfant, leur localisation principale est rétropéritonéale. (source : Institut Gustave Roussy)
Références
- Fridman R, Giaccone G, Kanemoto T, et al. Reconstituted basement membrane (matrigel) and laminin can enhance the tumorigenicity and the drug resistance of small cell lung cancer cell lines. Proc Natl Acad Sci USA 1990 ; 87 : 6698–6702. [CrossRef] [Google Scholar]
- Damiano JS, Cress AE, Hazlehurst LA, et al. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999 ; 93 : 1658–1667. [PubMed] [Google Scholar]
- Park CC, Zhang HJ, Yao ES, et al. Beta1 integrin inhibition dramatically enhances radiotherapy efficacy in human breast cancer xenografts. Cancer Res 2008 ; 68 : 4398–4405. [CrossRef] [PubMed] [Google Scholar]
- Hsieh YT, Gang EJ, Geng H, et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood 2013 ; 121 : 1814–1818. [CrossRef] [PubMed] [Google Scholar]
- Hazlehurst LA, Damiano JS, Buyuksal I, et al. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene 2000 ; 19 : 4319–4327. [CrossRef] [PubMed] [Google Scholar]
- Shain KH, Landowski TH, Dalton WS. Adhesion-mediated intracellular redistribution of c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long confers resistance to CD95-induced apoptosis in hematopoietic cancer cell lines. J Immunol 2002 ; 168 : 2544–2553. [CrossRef] [PubMed] [Google Scholar]
- Mishra S, Zhang B, Cunnick JM, et al. Resistance to imatinib of bcr/abl p190 lymphoblastic leukemia cells. Cancer Res 2006 ; 66 : 5387–5393. [CrossRef] [PubMed] [Google Scholar]
- Mitsiades CS, Mitsiades NS, Munshi NC, et al. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: interplay of growth factors, their receptors and stromal interactions. Eur J Cancer 2006 ; 42 : 1564–1573. [CrossRef] [PubMed] [Google Scholar]
- Shain KH, Yarde DN, Meads MB, et al. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: implications for microenvironment influence on tumor survival and proliferation. Cancer Res 2009 ; 69 : 1009–1015. [CrossRef] [PubMed] [Google Scholar]
- Chao Y, Wu Q, Shepard C, Wells A. Hepatocyte induced re-expression of E-cadherin in breast and prostate cancer cells increases chemoresistance. Clin Exp Metastasis 2012 ; 29 : 39–50. [CrossRef] [PubMed] [Google Scholar]
- Parmar A, Marz S, Rushton S, et al. Stromal niche cells protect early leukemic FLT3-ITD+ progenitor cells against first-generation FLT3 tyrosine kinase inhibitors. Cancer Res 2011 ; 71 : 4696–4706. [CrossRef] [PubMed] [Google Scholar]
- Sohara Y, Shimada H, Minkin C, et al. Bone marrow mesenchymal stem cells provide an alternate pathway of osteoclast activation and bone destruction by cancer cells. Cancer Res 2005 ; 65 : 1129–1135. [CrossRef] [PubMed] [Google Scholar]
- Ara T, Song L, Shimada H, et al. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res 2009 ; 69 : 329–337. [CrossRef] [PubMed] [Google Scholar]
- Perez LE, Parquet N, Shain K, et al. Bone marrow stroma confers resistance to Apo2 ligand/TRAIL in multiple myeloma in part by regulating c-FLIP. J Immunol 2008 ; 180 : 1545–1555. [CrossRef] [PubMed] [Google Scholar]
- Ara T, Nakata R, Sheard MA, et al. Critical role of STAT3 in IL-6-mediated drug resistance in human neuroblastoma. Cancer Res 2013 ; 73 : 3852–3864. [CrossRef] [PubMed] [Google Scholar]
- Duda DG, Kozin SV, Kirkpatrick ND, et al. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies?. Clin Cancer Res 2011 ; 17 : 2074–2080. [CrossRef] [PubMed] [Google Scholar]
- Hattori K, Heissig B, Rafii S. The regulation of hematopoietic stem cell and progenitor mobilization by chemokine SDF-1. Leuk Lymphoma 2003 ; 44 : 575–582. [CrossRef] [PubMed] [Google Scholar]
- Shiozawa Y, Pedersen EA, Havens AM, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest 2011 ; 121 : 1298–1312. [CrossRef] [PubMed] [Google Scholar]
- Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer 2009 ; 9 : 665–674. [CrossRef] [PubMed] [Google Scholar]
- Weekes CD, Song D, Arcaroli J, et al. Stromal cell-derived factor 1alpha mediates resistance to mTOR-directed therapy in pancreatic cancer. Neoplasia 2012 ; 14 : 690–701. [PubMed] [Google Scholar]
- Hartmann TN, Burger JA, Glodek A, et al. CXCR4 chemokine receptor and integrin signaling co-operate in mediating adhesion and chemoresistance in small cell lung cancer (SCLC) cells. Oncogene 2005 ; 24 : 4462–4471. [CrossRef] [PubMed] [Google Scholar]
- Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010 ; 29 : 4741–4751. [CrossRef] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011 ; 10 : 417–427. [Google Scholar]
- Bisping G, Leo R, Wenning D, et al. Paracrine interactions of basic fibroblast growth factor and interleukin-6 in multiple myeloma. Blood 2003 ; 101 : 2775–2783. [CrossRef] [PubMed] [Google Scholar]
- Nefedova Y, Landowski TH, Dalton WS. Bone marrow stromal-derived soluble factors and direct cell contact contribute to de novo drug resistance of myeloma cells by distinct mechanisms. Leukemia 2003 ; 17 : 1175–1182. [CrossRef] [PubMed] [Google Scholar]
- Silverman AM, Nakata R, Shimada H, et al. A galectin-3-dependent pathway upregulates interleukin-6 in the microenvironment of human neuroblastoma. Cancer Res 2012 ; 72 : 2228–2238. [CrossRef] [PubMed] [Google Scholar]
- Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol 2002 ; 2 : 569–579. [PubMed] [Google Scholar]
- Andre F, Schartz NE, Chaput N, et al. Tumor-derived exosomes: a new source of tumor rejection antigens. Vaccine 2002 ; 20 suppl 4 : A28–A31. [CrossRef] [PubMed] [Google Scholar]
- Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 2013 ; 339 : 286–291. [CrossRef] [PubMed] [Google Scholar]
- Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol 2011 ; 21 : 139–146. [CrossRef] [PubMed] [Google Scholar]
- Fabbri M, Paone A, Calore F, et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci USA 2012 ; 109 : E2110–E2116. [CrossRef] [Google Scholar]
- Park D, Sykes DB, Scadden DT. The hematopoietic stem cell niche. Front Biosci 2012 ; 17 : 30–39. [CrossRef] [Google Scholar]
- Fidler IJ. Seed and soil revisited: contribution of the organ microenvironment to cancer metastasis. Surg Oncol Clin N Am 2001 ; 10 : 257–269. [PubMed] [Google Scholar]
- Bidard FC, Poupon MF. Biologie du processus métastatique. Med Sci (Paris) 2012 ; 28 : 89–95. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J Clin Oncol 2010 ; 28 : 4022–4028. [CrossRef] [PubMed] [Google Scholar]
- Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012 ; 21 : 309–322. [CrossRef] [PubMed] [Google Scholar]
- Denardo DG, Brennan DJ, Rexhepaj E, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 2011 ; 1 : 54–67. [CrossRef] [PubMed] [Google Scholar]
- Multhoff G, Radons J., Radiation inflammation, immune responses in cancer. Front Oncol 2012 ; 2 : 58. [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005 ; 438 : 820–827. [CrossRef] [PubMed] [Google Scholar]
- Ferrara N. From the discovery of vascular endothelial growth factor to the introduction of avastin in clinical trials: an interview with Napoleone Ferrara by Domenico Ribatti. Int J Dev Biol 2011 ; 55 : 383–388. [CrossRef] [PubMed] [Google Scholar]
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008 ; 8 : 592–603. [CrossRef] [PubMed] [Google Scholar]
- Nabors LB, Mikkelsen T, Hegi ME, et al. A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306). Cancer 2012 ; 118 : 5601–5607. [CrossRef] [PubMed] [Google Scholar]
- Welschinger R, Liedtke F, Basnett J, et al. Plerixafor (AMD3100) induces prolonged mobilization of acute lymphoblastic leukemia cells and increases the proportion of cycling cells in the blood in mice. Exp Hematol 2013 ; 41 : 293–302. [CrossRef] [PubMed] [Google Scholar]
- Agarwal A, Fleischman AG, Petersen CL, et al. Effects of plerixafor in combination with BCR-ABL kinase inhibition in a murine model of CML. Blood 2012 ; 120 : 2658–2668. [CrossRef] [PubMed] [Google Scholar]
- Chomel JC, Aggoune D, Sorel N, Turhan AG. Leucémie myéloïde chronique : un modèle de dialogue entre la cellule souche leucémique et la niche hématopoïétique. Med Sci (Paris) 2014 ; 30 : 452–461. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Fridman WH, Sautès-Fridman C. Le microenvironnement tumoral : matrice nourricière, champ de bataille et cible thérapeutique des cancers. Med Sci (Paris) 2014 ; 30 : 359–365. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Provot S. Contrôle de la croissance et de la dissémination tumorales par le microenvironnement : certitudes et hypothèses émergentes. Med Sci (Paris) 2014 ; 30 : 366–371. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Fellouse FA. Les exosomes du stroma permettent au cellules cancéreuses de s’auto-activer. Med Sci (Paris) 2014 ; 30 : 405–407. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Bensimon J. Le switch angiogénique ou comment réveiller les cellules tumorales dormantes. Med Sci (Paris) 2012 ; 28 : 1069–1071. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Hubert S, Abastado JP. Les étapes précoces du processus métastatique. Med Sci (Paris) 2014 ; 30 : 378–384. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des tableaux
Agents thérapeutiques agissant sur le microenvironnement tumoral approuvés ou en cours d’étude clinique.
Liste des figures
|
Figure 1. Interactions entre cellules tumorales et cellules mésenchymateuses du stroma. Ces interactions et les voies de signalisation décrites favorisent la survie des cellules tumorales et la résistance aux thérapies anticancéreuses. EP : prostaglandin E2 receptor. BIM : membre de la famille Bcl2 proapoptotique ; FLIP : FLICE-like inhibitory protein (antiapoptotique) ; VEGF : vascular endothelial growth factor ; FGF : fibroblast growth factor ; PGE2 : prostaglandine E2. |
| Dans le texte | |
|
Figure 2. Contrôle de l’environnement tumoral et métastatique par la moelle osseuse. CSH : cellule souche hématopoïétique ; CSM : cellule souche mésenchymateuse ; PCE : précurseur endothélial ; CE : cellule endothéliale. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.