")
")
| Issue |
Med Sci (Paris)
Volume 29, Number 6-7, Juin–Juillet 2013
|
|
|---|---|---|
| Page(s) | 607 - 616 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2013296013 | |
| Published online | 12 juillet 2013 | |
Hypertension artérielle pulmonaire
Un parfum d’auto-immunité
Pulmonary arterial hypertension: a flavor of autoimmunity
1
Université Paris-Sud, faculté de médecine, hôpital de Bicêtre, 94270 Le Kremlin-Bicêtre, France
2
AP-HP, département hospitalo-universitaire thorax innovation, centre de référence de l’hypertension pulmonaire sévère, service de pneumologie et réanimation respiratoire, hôpital de Bicêtre, 94270 Le Kremlin-Bicêtre, France
3
Inserm UMR-S 999, Labex LERMIT, hypertension artérielle pulmonaire : physiopathologie et innovation thérapeutique, centre chirurgical Marie Lannelongue, 133, avenue de la Résistance, 92350 Le Plessis-Robinson, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Il est admis que l’auto-immunité résulte d’une combinaison de risques, tels qu’un terrain génétique, des facteurs environnementaux déclenchants et des événements stochastiques inconnus. L’hypertension artérielle pulmonaire (HTAP) partage avec les maladies auto-immunes prototypiques les facteurs de risque génétiques, la prédominance féminine et son corollaire d’influence des hormones sexuelles, l’association à d’autres maladies inflammatoires ou auto-immunes, le déficit des fonctions immunorégulatrices et la présence d’auto-anticorps. Quelques cas indiquant l’effet bénéfique de thérapies immunosuppressives ou anti-inflammatoires dans l’HTAP sont rapportés, ce qui permet de recentrer les mécanismes immunitaires dans la physiopathologie de la maladie. Nous discutons dans cette revue des connaissances actuelles sur les mécanismes auto-immuns mis en œuvre dans l’HTAP, notamment l’élaboration d’une réponse immunitaire locale au sein du tissu pulmonaire, à savoir une néogenèse lymphoïde pulmonaire. Une meilleure compréhension du rôle de l’auto-immunité dans le remodelage vasculaire pourrait permettre de développer des stratégies d’immunomodulation ciblées dans l’HTAP.
Abstract
It is admitted that autoimmunity results from a combination of risks such as genetic background, environmental triggers, and stochastic events. Pulmonary arterial hypertension (PAH) shares with the so-called prototypic autoimmune diseases, genetic risk factors, female predominance and sex hormone influence, association with other chronic inflammatory and autoimmune diseases, defects in regulatory T cells function, and presence of autoantibodies. Case reports have been published indicating the beneficial effect of some immunosuppressive and anti-inflammatory therapies in PAH, supporting the potential role of immune mechanisms in the pathophysiology of the disease. In this review, we discuss the current knowledge on autoimmune mechanisms operating in PAH, especially mounting a local autoimmune response inside the pulmonary tissue, namely pulmonary lymphoid neogenesis. A better understanding of the role of autoimmunity in pulmonary vascular remodelling may help develop targeted immunomodulatory strategies in PAH.
© 2013 médecine/sciences – Inserm

Vignette (Photo © Inserm-Daniel Caro).
L’hypertension artérielle pulmonaire
L’hypertension artérielle pulmonaire (HTAP) correspond à un groupe de maladies d’évolution progressive caractérisées par une augmentation chronique de la pression artérielle pulmonaire moyenne (≥ 25 mmHg) sans anomalies du cœur gauche (l’HTAP est précapillaire, c’est-à-dire que la pression artérielle pulmonaire d’occlusion est normale ≤ 15 mmHg) (Encadré 1). L’HTAP entraîne, dans un premier temps, une hypertrophie compensatrice du ventricule droit (VD), suivie d’une défaillance cardiaque droite dans la phase tardive et symptomatique du développement de cette pathologie sévère [1, 2]. L’HTAP peut être idiopathique (HTAPI), héritable ou familiale, induite par la prise de médicaments ou de toxiques, ou compliquer l’évolution de certaines maladies (HTAP associée à une connectivite, une cardiopathie congénitale, l’infection par le virus de l’immunodéficience humaine [VIH], une hypertension portale, etc.) [3]. La fréquence de l’HTAPI est de 5 à 15 cas/million de personnes, et de 50 cas par million tous sous-groupes confondus. Ces sous-groupes sont définis dans la classification officielle des hypertensions pulmonaires, classification qui stratifie les patients en fonction de la prise en charge thérapeutique. Des mutations germinales du gène BMPR2 (bone morphogenetic protein receptor type 2) sont détectées dans 10 à 40 % des HTAPI, et 58 à 74 % des HTAP familiales [4]. Les traitements actuels de l’HTAP reposent sur des vasodilatateurs ciblant des médiateurs du dysfonctionnement endothélial pulmonaire caractéristique de la maladie (prostacycline, antagonistes des récepteurs de l’endothéline-1, inhibiteurs des phosphodiestérases de type 5) [2, 5]. Malgré ces innovations thérapeutiques, l’HTAP reste une maladie très sévère dont la survie médiane est de l’ordre de 3 à 5 ans [6]. La transplantation pulmonaire ou cardio-pulmonaire est proposée aux patients au stade terminal de l’HTAP avec insuffisance cardiorespiratoire et réfractaires aux traitements médicaux [1].
L’HTAP est une maladie multifactorielle caractérisée par des atteintes structurelles et fonctionnelles du lit vasculaire pulmonaire qui conduisent à un remodelage intense des artérioles pulmonaires. Bien que les mécanismes exacts conduisant à ce remodelage ne soient pas complètement élucidés, des facteurs directement impliqués, en particulier le dysfonctionnement endothélial, la prolifération excessive des cellules musculaires lisses et des cellules endothéliales, la thrombose in situ et l’inflammation ont été identifiés ces dernières années [7]. Il est important de noter que les patients avec un désordre immunitaire associé ont des lésions indiscernables de celles rencontrées chez les patients avec une HTAPI, et répondent aux mêmes traitements, ce qui suggère des mécanismes effecteurs communs. Ces HTAP sont caractérisées par la présence de lésions plexiformes, composées de cellules endothéliales activées en prolifération présentant beaucoup de caractéristiques des cellules cancéreuses. Les cellules inflammatoires sont présentes autour et dans les lésions plexiformes, suggérant un rôle de l’inflammation dans la pathogenèse de cette maladie. De nombreuses observations appuient cette hypothèse. Des concentrations sériques élevées de cytokines pro-inflammatoires et de chimiokines (cytokines chimioattractantes) ont été mesurées chez les patients souffrant d’HTAPI, c’est-à-dire sans autre maladie inflammatoire, infectieuse ou auto-immune avérée sous-jacente [7]. Les lésions vasculaires pulmonaires d’HTAPI sont également le siège d’une intense production de chimiokines associée à un recrutement parfois important de cellules inflammatoires [7]. Des auto-anticorps circulants, anti-cellules endothéliales et anti-fibroblastes notamment, sont également retrouvés chez 10 à 40 % des patients souffrants d’HTAPI [8–10], signant peut-être la présence de mécanismes auto-immuns liés à l’activation du complément dans la genèse des lésions vasculaires pulmonaires d’HTAP. Enfin, l’implication de mécanismes immuns dans l’étiologie de l’HTAP semble évidente dans les HTAP associées à des maladies auto-immunes ou à l’infection par le VIH [7], où l’HTAP se développe sur un terrain immunitaire altéré. Dans ce contexte, des traitements immunosuppresseurs ou des corticostéroïdes peuvent significativement améliorer les paramètres hémodynamiques et cliniques de ces patients [11], soulignant le rôle de phénomènes immuns dans le développement et l’entretien de ces HTAP
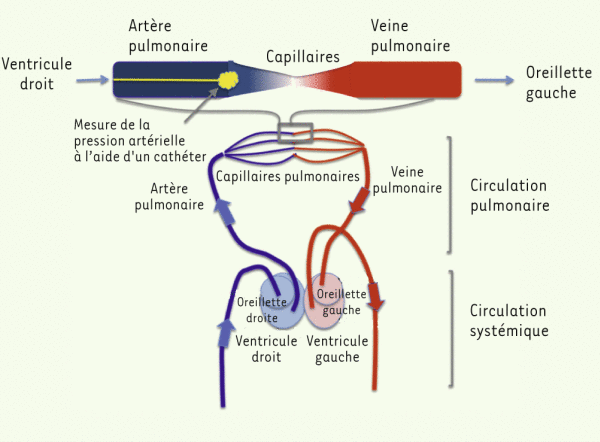
1 Caractéristiques des HTAP pré et postcapillaires
Au cours du cathétérisme cardiaque droit, on mesure les pressions et les débits au niveau des artères pulmonaires et du cœur droit. On peut aussi estimer de façon indirecte la pression de l’oreillette gauche en mesurant la pression artérielle pulmonaire occluse (en bloquant le cathéter ou en gonflant un ballonnet situé au bout du cathéter). En fonction des valeurs de la pression artérielle pulmonaire occluse, les hypertensions pulmonaires sont classées en hypertensions pulmonaires précapillaires ou postcapillaires (voir schéma ci-dessous). Les hypertensions pulmonaires précapillaires se définissent par une pression artérielle pulmonaire occluse normale, inférieure ou égale à 15 mmHg (on parle alors d’hypertension « artérielle » pulmonaire). Cela traduit en général une maladie du réseau artériel pulmonaire. Les hypertensions pulmonaires postcapillaires (on parle alors d’hypertension « veineuse » pulmonaire) se caractérisent par une pression artérielle pulmonaire occluse élevée, supérieure à 15 mmHg. Cette valeur traduit en général une maladie du cœur gauche. L’hypertension pulmonaire postcapillaire est passive, sans maladie à proprement parler du réseau artériel pulmonaire qui est engorgé à cause de l’insuffisance cardiaque gauche.
Paradigmes de l’auto-immunité dans l’HTAP
Prédominance féminine
Les données épidémiologiques indiquent une nette prédominance féminine chez les patients avec HTAP, avec un rapport de 1,9 à 4,1 femmes pour 1 homme dans l’HTAPI [12], et un rapport de 2,4/1 dans l’HTAP héritable [4]. Au cours des HTAP associées à d’autres maladies, le rapport femme/homme est influencé par les caractéristiques épidémiologiques de la maladie prédisposant à l’HTAP. La raison d’une plus forte prévalence de l’HTAP chez les femmes n’est pas claire. Certaines hypothèses avancent le rôle des hormones sexuelles (œstrogènes), de l’auto-immunité, ou d’un locus lié à l’X dans la prédisposition à la maladie [13]. La théorie hormonale, qui stipule que les œstrogènes orientent vers une réponse immunitaire CD4 de type Th2, avec pour conséquence une plus forte activation des cellules B et une augmentation de la production d’anticorps est l’observation la plus convaincante pour expliquer le biais de fréquence des affections auto-immunes en faveur des femmes. À noter que les androgènes favorisent le développement de réponses immunitaires de type Th1 et l’activation de cellules CD8+.
On peut se poser la question inverse : pourquoi les maladies auto-immunes (et l’HTAP) sont-elles moins prévalentes chez les hommes, et peut-il exister des facteurs protecteurs, y compris vasoprotecteurs et cardioprotecteurs ? Chez les patients atteints d’HTAP héritable et présentant une mutation du gène BMPR2, la pénétrance de la mutation a été récemment estimée à 42 % chez les femmes et seulement 14 % chez les hommes [14]. Il faut cependant souligner que les manifestations cliniques ne sont pas différentes chez les hommes et les femmes. Cependant, les hommes ont une évolution clinique plus sévère et un risque accru de mortalité. Ceci pourrait être lié au rôle paradoxal des œstrogènes dans l’HTAP, qui favorisent potentiellement l’auto-immunité mais possèdent également un rôle protecteur vasculaire [15]. Une possible influence des hormones sexuelles sur le développement et l’expression clinique de la maladie est aussi suggérée par la possible apparition des signes de l’HTAP durant la grossesse, ou peu de temps avant l’accouchement. Bien que les mécanismes mis en jeu restent à définir, ces observations vont dans le sens de la prédominance féminine généralement observée dans les maladies auto-immunes [13].
Association entre HTAP et maladies inflammatoires, auto-immunes ou infectieuses
Il est reconnu depuis presque 50 ans qu’il existe des associations ou comorbidités entre HTAP et désordres auto-immuns (lupus érythémateux disséminé, syndrome de Sjögren, sclérodermie, thyroïdite, etc.) ou infections chroniques (VIH, bilharziose, HHV8 [virus de l’herpès humain de type 8]) pouvant conduire à une immunodéficience [1, 3]. Un point commun à ces affections immunitaires est l’immunodéficience avérée ou latente, qui pourrait conduire à une dérégulation du système immunitaire et à l’activation de cellules T et B pathogéniques. Mais le rôle de l’auto-imunité dans la pathogenèse de l’HTAP est mal compris, et rien n’indique si cette auto-immunité est la cause ou la conséquence de l’HTAP. Il est clair que des cellules T et B sont présentes dans les lésions vasculaires pulmonaires, et que des cellules dendritiques envahissent les lésions vasculaires aussi bien dans l’HTAPI humaine que l’HTAP expérimentale [7]. Des dépôts d’immunoglobulines G (IgG) ont même été détectés dans et autour des lésions plexiformes de patients avec HTAPI [16, 17]. Les effecteurs d’une réponse immunitaire locale sont donc présents au niveau des vaisseaux remodelés de patients souffrant d’HTAPI.
Défauts des cellules T régulatrices
L’HTAP est associée à des défauts du compartiment cellulaire CD4+, qui se traduisent par un déficit en cellules CD4+, une diminution du rapport CD4/CD8, et/ou une diminution du pourcentage de cellules CD4+CD25+, la sous-population contenant des cellules T régulatrices (Treg) potentielles [16]. La déplétion physique ou fonctionnelle des Treg, qui se traduit par un déséquilibre entre cellules effectrices et régulatrices, est un évènement démontré comme central dans le développement des maladies auto-immunes et suffit à rompre la tolérance au soi conduisant aux maladies auto-immunes chroniques. Dans le sang périphérique, des études contradictoires ont rapporté, soit une augmentation des Treg [23, 24], soit un pourcentage inchangé de cellules Treg mais dont la fonction est altérée, en lien avec une surexpression de leptine [25]. Dans le poumon, il a été montré une diminution du nombre de Treg Foxp3+ périvasculaires [26]. Il a été récemment démontré que le blocage du récepteur du VEGF (vascular endothelial growth factor) (VEGFR2) induit une apoptose significative des cellules endothéliales pulmonaires chez les rats déficients en cellules T, mais pas chez ces rats après une reconstitution immunitaire. Des transferts de cellules Treg avant induction de l’agression endothéliale suggèrent que les Treg fonctionnent en limitant l’agression endothéliale, et pourraient jouer un rôle protecteur vis-à-vis du développement de l’HTAP, en plus de leur action régulatrice éventuelle d’une réponse auto-immune [27].
Il est intéressant de noter que la voie BMPR-II qui est altérée au cours de l’HTAP héritable, mais aussi dans d’autres formes de la maladie, joue un rôle dans le développement des cellules T dans le thymus [16], ce qui pourrait contribuer à un défaut intrinsèque de la fonction et/ou du nombre de Treg chez les patients souffrant d’HTAP. Bien que ces études ouvrent de nouvelles hypothèses dans la physiopathologie de l’HTAP, la fonction régulatrice suppressive de ces cellules n’a pas encore été explorée, et l’identification des Treg par ses marqueurs reste à redéfinir [28].
Des auto-anticorps ciblant la paroi vasculaire
La recherche d’auto-anticorps chez les patients avec HTAPI ou avec une maladie auto-immune associée, et plus particulièrement la sclérodermie, focalise de plus en plus l’attention. On estime à 30-40 % les patients avec HTAPI qui ont des anticorps antinucléaires, et parmi ces patients, à 10-15 % ceux qui ont des anticorps anti-phospholipides [29]. Les anticorps anti-phospholipides sont capables de se lier aux cellules endothéliales et de les activer [30]. Les IgG de patients avec HTAP et/ou sclérodermie se lient aux cellules musculaires lisses et induisent la contraction cellulaire [31]. Des anticorps anti-cellules endothéliales sont détectés dans les maladies auto-immunes associées à l’HTAP, comme le lupus et la sclérodermie [32, 33]. Dans les HTAP associées au lupus et au syndrome de Sjögren, des dépôts d’anticorps et de molécules du complément sont trouvés dans les parois vasculaires [30]. L’augmentation locale de la production d’IL-1 (interleukine-1) et d’IL-6 [34], deux cytokines pro-inflammatoires qui interviennent dans l’activation, la prolifération et la différenciation des cellules B, ainsi que la présence de mastocytes dans et autour des lésions vasculaires [35], sont des éléments favorables à cette auto-immunité locale. Les mastocytes sont connus comme source d’IL-4 pour l’expansion locale des cellules B et font le lien entre la réponse immunitaire innée et adaptative, notamment dans un contexte d’auto-immunité [36].
Plus récemment, des recherches systématiques d’auto-anticorps de type IgG contre différents composants de la paroi vasculaire (cellules endothéliales, mais aussi cellules musculaires lisses et fibroblastes) ont été entreprises, ainsi que la recherche des cibles de ces auto-anticorps par des approches de protéomique, avec l’objectif d’identifier des biomarqueurs pour le diagnostic et le suivi clinique de l’HTAP [8–10]. Parmi les 21 cibles reconnues dans les fibroblastes figurent des acteurs jouant un rôle clé dans le maintien de l’homéostasie [10]. Cependant, parmi toutes les cibles identifiées, il reste à définir lesquelles sont les cibles d’auto-anticorps réellement pathogènes, qui puissent influencer la fonction vasculaire et/ou jouer un rôle dans le remodelage. Il faut noter que l’analyse différentielle réalisée par cette approche de protéomique en gels bidimensionnels reflète les modifications physiopathologiques des différents types cellulaires mis en jeu dans le remodelage, mais ne favorise pas la détection de cibles membranaires. Ceci ne remet pas en cause cependant le rôle pathogène potentiel d’auto-anticorps dirigés contre des composants cytoplasmiques ou nucléaires, suite à une agression endothéliale initiale de type toxique, induisant l’apoptose endothéliale et l’exposition d’antigènes cryptiques ou de néoantigènes. Une étude récente a cependant confirmé la prévalence d’anticorps anti-cellules endothéliales reconnaissant cette fois des composants de la surface cellulaire chez les patients atteints d’HTAPI (prévalence 62 %) ou d’HTAP associée (prévalence 78 %), comparés aux témoins (prévalence 32 %) [37]. La présence de ces auto-anticorps indique la possibilité d’un mécanisme humoral dans la pathogenèse de l’HTAP. En conclusion, la prévalence augmentée chez les patients HTAP par rapport aux témoins, les différences quantitatives entre patients et témoins, la sous-classe prépondérante IgG, et dans certaines études l’effet fonctionnel des anticorps en relation avec la pathogénicité sont autant d’arguments qui vont dans le sens de la notion d’auto-anticorps pathologiques dans l’HTAP, par opposition aux anticorps naturels.
Critères manquants pour valider l’hypothèse auto-immune dans l’HTAP
La présence d’auto-anticorps chez les patients HTAP indique un possible mécanisme humoral dans la pathogenèse de la maladie, avec comme hypothèse la présence, dans les sérums de patients, d’auto-anticorps impliqués dans le dysfonctionnement endothélial pulmonaire et participant au remodelage vasculaire. Les dépôts d’Ig observés au niveau vasculaire pulmonaire pourraient refléter de tels mécanismes [16, 17]. Cependant, cela reste pour le moment une observation indirecte associée à la maladie et ne constitue en aucun cas une preuve d’auto-immunité, pour laquelle une démonstration directe de la pathogénicité est requise, d’une part in vivo dans les modèles expérimentaux et, d’autre part, par transfert de sérum ou de cellules de l’homme à l’animal, selon les critères toujours reconnus de Rose et Bona [38]. Selon ces auteurs, trois types de preuves doivent être réunies pour établir l’origine auto-immune d’une maladie humaine : (1) une preuve directe du transfert de la maladie par des cellules T ou des anticorps pathogènes ; (2) des preuves indirectes basées sur l’induction de la maladie dans des modèles expérimentaux ; (3) des preuves indirectes apportées par des arguments cliniques. Les preuves indirectes s’accumulent et certaines équipes, dont la nôtre, s’attèlent à montrer des preuves directes. En effet, on commence à montrer que des auto-anticorps présents dans un modèle expérimental d’HTAP sont capables de transférer la maladie à l’animal [39], et nous envisageons des expériences de transfert de sérum de patient à l’animal.
Follicules lymphoïdes tertiaires périvasculaires : témoins d’une réponse (auto) immune locale ?
Des structures lymphoïdes organisées appelées tissus lymphoïdes tertiaires, qui ressemblent aux organes secondaires lymphoïdes, ont été très fréquemment observées dans les tissus qui sont les cibles d’un processus inflammatoire chronique comme l’auto-immunité et l’infection. Cette observation a suggéré que la néogenèse lymphoïde pourrait intervenir dans le maintien des réponses immunitaires contre des antigènes persistants, en particulier des auto-antigènes [41]. Cependant, malgré la démonstration que les structures lymphoïdes tertiaires sont actives immunologiquement, il est difficile de déterminer si c’est leur formation qui conduit à la maladie ou si c’est l’auto-immunité qui induit leur développement [41]. Des signes de mobilisation du système immunitaire ont été très récemment identifiés par notre équipe dans les poumons de patients avec HTAPI [17], et des follicules tertiaires ont été caractérisés à proximité des vaisseaux remodelés. La fréquence de ces structures a été évaluée dans des poumons explantés de patients souffrant d’HTAPI, des poumons témoins et des poumons d’HTAP induite par un hyperdébit (cardiopathie congénitale avec shunt gauche-droit, représentant un contrôle d’HTAP d’origine connue et a priori non immunologique). Contrairement aux poumons des témoins et des patients souffrants de cardiopathie congénitale avec HTAP, les poumons de patients atteints d’HTAPI contiennent des follicules tertiaires périvasculaires, divisés en aires T et B alimentées par des vaisseaux à cellules endothéliales dites « hautes » (high endothelial venules)1, et comportant des cellules dendritiques (Figure 1). Ces structures pourraient se mettre en place sous l’influence de cytokines/chimiokines intervenant dans l’organogenèse des structures lymphoïdes et dont la surexpression a été également mise en évidence au niveau pulmonaire dans l’HTAPI, et en particulier localisée au niveau des follicules lymphoïdes : lymphotoxine-α/-β, CCL19, CCL20, CCL21 et CXCL13. Cette expression ectopique pulmonaire pourrait expliquer la déplétion au niveau circulant des lymphocytes exprimant CCR6 et CXCR5 (les récepteurs de CCL20 et CXCL13)2. La présence de facteurs de survie tels que l’IL-7 et le PDGF-A (plateled-derived growth factor-A) indique que ces follicules tertiaires pourraient perdurer. Ces structures sont connectées aux artères remodelées par un réseau de cellules stromales ER-TR7+ et drainées par des vaisseaux lymphatiques dilatés. La présence de centres germinatifs, de cellules dendritiques folliculaires et de lymphocytes T helper folliculaires (TFH), la surexpression de l’enzyme AID (activation-induced cytidine deaminase) et du facteur de transcription Bcl6 (tous deux nécessaires aux mécanismes de diversification des immunoglobulines [Ig]), couplées à l’observation d’une accumulation de plasmocytes CD138+ autour des vaisseaux remodelés dans des zones de dépôt d’Ig, sont en faveur d’une production d’anticorps et d’une commutation de classe isotypique locale d’anticorps potentiellement pathogènes.
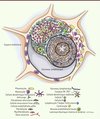 |
Figure 1. Néogenèse lymphoïde dans les lésions vasculaires pulmonaires de patients avec HTAP idiopathique. Nous émettons l’hypothèse que les auto-anticorps dirigés contre les fibroblastes et les cellules endothéliales fréquemment trouvés dans le sérum des patients émergent suite à la sélection positive et à l’activation de cellules B autoréactives au sein des follicules lymphoïdes tertiaires. Ces cellules B se développent dans l’adventice des petites artères pulmonaires remodelées dans le poumon des patients avec HTAP, fournissant ainsi un microenvironnement optimal pour la génération d’auto-anticorps pathogènes. À l’image des follicules lymphoïdes secondaires trouvés dans les ganglions, ces follicules lymphoïdes tertiaires sont organisés en zones T et B distinctes avec des centres germinatifs Bcl2-Ki-67+. Des antigènes du soi dérivés de composants vasculaires pourraient atteindre ces follicules lymphoïdes tertiaires par les conduits ER-TR7+ formés par des fibroblastes réticulaires et par des vaisseaux lymphatiques périvasculaires dilatés, et pourraient être présentés aux cellules T et B autoréactives par des cellules dendritiques myéloïdes. Les cellules dendritiques folliculaires exposent les complexes immuns à leur surface et activent les lymphocytes B. Les cellules inflammatoires constituant les follicules lymphoïdes tertiaires pourraient provenir des vasa vasorum se développant dans l’adventice des artères pulmonaires remodelées. Les cellules T naïves, pourraient également entrer dans les structures lymphoïdes tertiaires via des HEV (high endothelial venules) très différenciées, et être polarisées vers le phénotype Th17 favorable à l’induction de l’autoimmunité. De plus les cellules T helper folliculaires pourraient apporter l’aide nécessaire aux cellules B des centres germinatifs (centrocytes). Il en résulterait des hypermutations somatiques, des recombinaisons pour la commutation de classe et la sélection de cellules B de haute affinité. Les vaisseaux lymphatiques afférents dilatés et le réseau de vasa vasorum pourraient apporter aux structures lymphoïdes tertiaires les (auto)antigènes pulmonaires, les cellules dendritiques chargées en (auto)antigènes et les cellules T mémoires (extrait de [17], avec l’autorisation de © Am J Respir Crit Care Med). |
Tous ces éléments suggèrent la mise en place d’une néogenèse lymphoïde pulmonaire spécifique de l’HTAPI, ce qui est un argument nouveau en faveur de mécanismes immunologiques dans cette maladie vasculaire sévère. Ces structures lymphoïdes tertiaires et leur environnement pourraient être le siège de la génération et de la maturation d’effecteurs immunitaires, notamment des autoanticorps [41], comme on les rencontre dans les organes lymphoïdes secondaires. Cette découverte ouvre un nouveau paradigme en faveur de l’auto-immunité dans la physiopathologie de l’HTAP. Elle permet d’envisager une réponse immunitaire locale focalisée qui devrait laisser son empreinte dans les répertoires T et B périphériques qui sont en cours d’analyse. Une meilleure compréhension du rôle de la néogenèse lymphoïde dans la promotion de l’inflammation chronique pourrait conduire à de nouvelles stratégies thérapeutiques ciblant les processus immunopathologiques dans l’HTAP.
Vers une signature immunitaire chez les patients HTAP
Les follicules lymphoïdes périvasculaires observés dans les poumons de patients HTAP résultent clairement d’une néogenèse lymphoïde [17]. Il est tentant de penser que ce processus de néogenèse lymphoïde est lié au développement de l’HTAP et à la réponse contre des auto-antigènes vasculaires. Quels que soient ces auto-antigènes et les réponses T et B des cellules composant ces follicules (qu’il reste à définir précisément), l’analyse des répertoires des cellules T et B composant ces follicules pourrait être très informative. En effet un répertoire oligoclonal pourrait suggérer que ces follicules jouent un rôle dans une réponse immunitaire locale spécifique d’antigène. Nous analysons actuellement la diversité combinatoire des follicules lymphoïdes périvasculaires pulmonaires après leur microdissection laser par une analyse globale au niveau génomique (pour plus de détails sur l’approche, voir ImmunID Technologies, http://www.immunid.com/). Nous espérons ainsi définir une signature immunitaire de l’HTAP, que nous rechercherons ensuite dans les populations T et B circulantes. Cette signature immunitaire, si elle est détectable en périphérie, pourrait constituer un biomarqueur identifiant les patients candidats à une immuno-intervention, mais aussi un marqueur diagnostique non invasif et/ou un marqueur pronostique de l’activité, du stade clinique ou du sous-groupe de la maladie, utilisable en pratique clinique.
L’immunothérapie : vers de nouvelles stratégies thérapeutiques dans l’HTAP ?
Les thérapies actuelles de l’HTAP ont essentiellement pour but de réduire les résistances vasculaires pulmonaires en favorisant la vasodilatation pulmonaire (analogues des prostacyclines, inhibiteurs des phosphodiestérases de type 5 et antagonistes des récepteurs de l’endothéline) [2]. Ces thérapies ont permis d’atténuer les symptômes et d’améliorer la qualité de vie des personnes atteintes d’HTAP. Cependant, les résultats des traitements actuels sont insuffisants et de nouveaux traitements interférant avec les mécanismes physiopathologiques de l’HTAP à plusieurs niveaux sont nécessaires pour bloquer la progression de la maladie. La manifestation d’une inflammation, et plus spécifiquement d’une réponse immunitaire adaptative caractérisée par une néogenèse lymphoïde pulmonaire chez les patients atteints d’HTAP, laisse supposer que de nouvelles voies thérapeutiques visant la composante immunologique de cette maladie pourraient être envisagées.
Bien que les traitements actuels de l’HTAP aient tous des propriétés immunomodulatrices en plus de leurs propriétés vasodilatatrices [7], il n’y a aucune thérapie approuvée destinée à moduler spécifiquement les processus inflammatoires pour traiter l’HTAP. Cependant, la littérature fait état de cas isolés indiquant des effets potentiellement bénéfiques. Nous analysons ici quelques exemples sélectionnés.
Corticostéroïdes
Les corticostéroïdes ont des effets anti-inflammatoires, immunosuppresseurs, et antiprolifératifs bien connus [42]. Leur efficacité dans l’HTAP associée au lupus érythémateux disséminé et aux connectivites mixtes (mais pas à la sclérodermie systémique) a déjà été rapportée. Cependant, une publication récente décrit un cas mal documenté d’HTAPI (sans connectivite associée) traité par la prednisolone pour soigner un purpura thrombocytopénique idiopathique et dont certains paramètres échocardiographiques et cliniques ont été améliorés [43]. Cette amélioration a été incomplète et l’arrêt du traitement a entraîné une détérioration clinique de l’HTAP conduisant à la transplantation pulmonaire. Il est donc possible que certaines formes d’HTAP (qu’il faudra mieux caractériser par des biomarqueurs) puissent bénéficier d’une corticothérapie.
Anticorps anti-CD20
L’HTAP est une complication rare mais particulièrement grave du lupus érythémateux disséminé. Elle est signalée chez 0,5 % à 14 % de ces patients. Chez un patient souffrant de lupus érythémateux disséminé ayant développé une HTAP progressive et sévère au cours de sa maladie, résistante aux agents thérapeutiques spécifiques de l’HTAP et à de hautes doses de corticostéroïdes, l’utilisation de rituximab a permis une amélioration hémodynamique et clinique importante [44, et 56]. Le rituximab (Mabthéra™ ou Rituxan™) est un anticorps chimérique murin humanisé contre l’antigène CD20 [57]. Il est actif contre les cellules malignes exprimant l’antigène CD20, c’est-à-dire les lymphomes folliculaires de stade III-IV et les lymphomes non hodgkiniens agressifs diffus à grandes cellules B CD20+ [45]. D’une manière générale, il cible la plupart des lymphocytes B. Il est efficace dans de nombreuses maladies auto-immunes comme le lupus érythémateux avec glomérulonéphrite, le syndrome des anticorps antiphospholipides, certaines vascularites, etc. [44].
Les mécanismes physiopathologiques responsables de l’HTAP dans le lupus érythémateux disséminé ne sont pas bien identifiés. Bien qu’une atteinte directe due à des facteurs immunologiques n’ait pas été clairement démontrée, la détection d’anticorps antinucléaires circulants et de dépôts d’immunoglobulines et de molécules du complément dans la paroi des artères pulmonaires de ces patients [16, 17] sont en faveur d’une implication du système immunitaire dans la pathogenèse de l’HTAP.
Le rituximab induit la déplétion ciblée des lignées cellulaires exprimant l’antigène CD20 ce qui, chez les patients souffrant de lupus érythémateux disséminé, réduit l’activité de la maladie en diminuant le nombre de lymphocytes B autoréactifs dans les tissus lymphoïdes périphériques. Ceci s’accompagne souvent d’une réduction des taux circulants d’auto-anticorps caractéristiques du lupus érythémateux disséminé, ce qui, vraisemblablement, réduit la formation de complexes immuns et l’activation du complément qui pourraient être directement cytotoxiques. De plus, le rituximab pourrait abaisser la quantité de complexes immuns contenant des auto-anticorps qui, autrement, pourraient stimuler de nombreuses cellules de l’immunité innée (cellules dendritiques et macrophages) à produire et libérer des cytokines pro-inflammatoires. Les effets du rituximab pourraient également être indépendants de la production d’anticorps, le traitement diminuant la disponibilité en cellules B pour fournir des signaux de costimulation et présenter des (auto-)antigènes aux lymphocytes T. Il a été montré que le rituximab normalise l’expression des molécules de costimulation (CD40 et CD80) sur les lymphocytes B résiduels [46]. Des réductions similaires des ligands du CD40 (CD40L/CD154) ont été observées sur les lymphocytes T[47]. Ceci pourrait interrompre le cycle d’activation des cellules T autoréactives. L’élimination des lymphocytes B activés pourrait également diminuer les niveaux de cytokines et de chimiokines qu’ils produisent et qui sont capables de changer l’environnement local en milieu pro-inflammatoire. Le rituximab pourrait donc inhiber les processus d’induction et de perpétuation de la néogenèse lymphoïde pulmonaire, mais aussi de façon plus globale, l’inflammation chronique systémique et pulmonaire détectée dans l’HTAPI. Les cas d’HTAP associée ou idiopathique traités par rituximab devraient donc être étudiés avec soin pour rechercher d’éventuels effets positifs de cette molécule sur l’évolution de l’HTAP. De telles observations pourraient constituer un signal positif pour envisager l’utilisation du rituximab dans l’HTAP. Un essai est actuellement en cours dans l’HTAP de la sclérodermie (ClinicalTrials.gov Identifier : NCT01086540).
Anticorps anti-récepteur de l’IL-6
Les cytokines sont essentielles pour orchestrer la réponse immunitaire. Dans l’auto-immunité, le dérèglement du réseau des cytokines contribue aux atteintes tissulaires induites par l’inflammation, non seulement en les provoquant, mais aussi en les entretenant, ce qui favorise la progression vers une maladie chronique. La cytokine pro-inflammatoire IL-6 joue un rôle central dans ces processus, comme le montrent les modèles expérimentaux d’auto-immunité et les essais cliniques au cours desquels sa neutralisation a permis une amélioration de patients souffrant de la maladie de Castleman3 et de polyarthrite rhumatoïde [48]. L’IL-6 est indispensable au développement de la réponse immunitaire et, en particulier, de l’inflammation dépendante des lymphocytes T [48]. En effet, les activités biologiques de l’IL-6 incluent, entre autres, la différenciation des cellules B et des monocytes/macrophages et l’activation des cellules T. L’IL-6 joue même un rôle central dans des mécanismes clés de la réponse immunitaire adaptative : génération de centres germinatifs, hypermutations somatiques, maturation d’affinité des immunoglobulines, et différenciation des plasmocytes. Dans les modèles animaux de maladies auto-immunes, l’IL-6 a un rôle dans la génération de la réponse auto-immune T (et en particulier Th17) et dans la réponse humorale. Ainsi, l’IL-6 agit sur une grande variété de cellules, incluant les cellules immunocompétentes et les cellules hématopoïétiques, pour induire leur prolifération et leur différenciation [48]. L’hyperproduction de cette cytokine est une constante dans de nombreuses maladies auto-immunes, comme la polyarthrite rhumatoïde, le lupus érythémateux disséminé, la maladie de Castleman, l’arthrite idiopathique juvénile systémique (AIJS), et elle est responsable de nombreux symptômes cliniques associés à ces maladies [48]. C’est sur la base de ces observations qu’a été développé le tocilizumab (RoActemra™), un anticorps inhibiteur des récepteurs de l’IL-6, pour le traitement de la polyarthrite rhumatoïde, l’AIJS et la maladie de Castleman notamment [49].
Dans l’HTAP, l’IL-6 semble jouer un rôle important et pourrait également être une cible thérapeutique prometteuse. En effet, cette cytokine est suspectée d’intervenir dans la prolifération des cellules endothéliales et musculaires lisses des lésions d’HTAP [50]. On retrouve des niveaux élevés de cette cytokine dans le sérum des patients souffrant d’HTAP [34], et les concentrations d’IL-6 élevées semblent être associées à un pronostic plus médiocre, sans corrélation avec les données hémodynamiques de ces patients [51]. Ces observations révèlent vraisemblablement des marqueurs indépendants de la fonction cardiaque droite et potentiellement impliqués dans la physiopathologie de l’HTAP. Une étude récente a montré qu’un traitement par bosentan (un antagoniste des récepteurs de l’endothéline) exerce un effet anti-inflammatoire par une diminution des taux plasmatiques d’ICAM-1 (intercellular adhesion molecule-1) et d’IL-6, corrélée à une amélioration hémodynamique [52]. Enfin, une hypertension pulmonaire est également obtenue chez des souris transgéniques qui surexpriment l’IL-6 spécifiquement au niveau pulmonaire [50].
2 Paradigmes de l’auto-immunité dans l’HTAP
-
Association entre maladies inflammatoires, infectieuses et auto-immunes avec l’HTAP
-
Rôle physiopathologique des auto-anticorps in vitro [30, 31]
-
Néogenèse lymphoïde périvasculaire : follicules tertiaires pulmonaires [17]
-
Transfert de la maladie de l’animal malade à l’animal sain [39]
Étapes décisives restant à explorer
-
Transfert de la maladie de l’homme à l’animal
-
Mécanismes physiopathologiques des auto-anticorps dans l’HTAP humaine
-
Signature immunitaire pulmonaire de l’HTAP
-
Biomarqueur(s) circulant(s) chez les patients candidats à une immunosuppression
Points clés
-
Les processus inflammatoires apparaissent jouer un rôle important dans le remodelage vasculaire caractéristique de l’HTAP et pourraient être des cibles thérapeutiques pour le traitement ou la prévention de l’HTAP
-
Les études précliniques chez l’animal montrent des résultats prometteurs, mais cela reste largement inexploré dans l’HTAP humaine, excepté pour certains cas d’HTAP associés à des maladies auto-immunes/inflammatoires qui ont été améliorés sous immunosuppression
-
Les mécanismes inflammatoires auto-immuns mis en jeu sont complexes et mal compris, et doivent prédominer dans la physiopathologie chez certains sous-groupes de patients qu’il reste à définir par la recherche de biomarqueurs
-
Une meilleure définition de ces mécanismes pourrait faire émerger de nouvelles cibles thérapeutiques potentielles qui sont attendues compte tenu de l’arsenal thérapeutique actuel qui ne cible que le dysfonctionnement endothélial reflété par un excès de vasoconstriction et de prolifération
À l’heure actuelle, il existe trois cas publiés d’HTAP traitées par tocilizumab : un cas d’HTAP associée à une connectivite [53], et deux cas d’HTAP associées à une maladie de Castleman [54, 55]. Dans tous les cas, le tocilizumab a été donné pour soigner la maladie inflammatoire ou auto-immune et a permis une amélioration très marquée des paramètres cliniques et hémodynamiques des HTAP associées. Il a même apporté un bénéfice évident chez un des patients sévèrement atteint pourtant déjà traité avec une association des trois classes thérapeutiques de l’HTAP (prostacycline, bosentan et sildenafil) [54]. Ces trois malades présentaient des taux élevés d’IL-6 circulants. Le tocilizumab pourrait donc apporter un bénéfice thérapeutique aux HTAP caractérisées par des taux élevés de cette cytokine, comme les HTAP associées aux connectivites et, peut-être, à certains sous-groupes d’HTAPI.
Conclusion et perspectives
Avec la reconnaissance du rôle important de l’inflammation, voire de l’auto-immunité, dans le développement de l’HTAP, des traitements anti-inflammatoires/immunosuppresseurs et immunomodulateurs pourraient être proposés comme traitements ou adjuvants aux traitements existants de l’HTAP (voir Encadré 2). Les premiers essais, réalisés essentiellement dans quelques cas de maladies inflammatoires/auto-immunes associées à l’HTAP, semblent prometteurs, mais il est important et nécessaire de continuer à avancer dans la compréhension des mécanismes auto-immuns et inflammatoires dans l’HTAP, en particulier comprendre comment ils influencent le remodelage vasculaire, et de commencer à mettre en place des études cliniques contrôlées. Dans ce contexte la définition de biomarqueurs chez les patients HTAP candidats à une immunothérapie adjuvante serait d’une aide précieuse. Parmi ces biomarqueurs, la recherche d’une signature immunitaire chez les patients HTAP est une voie prometteuse.
Liens d’intérêt
Les auteurs Frédéric Perros, Marc Humbert et Sylvia Cohen-Kaminsky déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article. Dans les cinq années précédentes, Marc Humbert a participé à des essais cliniques et a conseillé des laboratoires travaillant dans le domaine de l’hypertension artérielle pulmonaire (Actelion, Aires, Bayer, GSK, Lilly, Novartis, Pfizer et United Therapeutics).
Remerciements
Nous remercions pour leur soutien la Fondation pour la recherche médicale (FRM DEQ20100318257), l’université Paris-Sud, l’Inserm, l’Assistance publique hôpitaux de Paris, le centre chirurgical Marie Lannelongue, la Chancellerie des universités de Paris, et l’association HTAP France. Frédéric Perros a été soutenu par la FRM en 2011 et 2012 (FRM DEQ20100318257).
« Le transit des lymphocytes naïfs du sang vers le tissu lymphoïde ganglionnaire Nécessite une étape de migration transendothéliale, qui prend place spécifiquement au sein des veinules postcapillaires appelées HEV. […] Les cellules endothéliales extrêmement différenciées qui tapissent ces vaisseaux HEV sont caractérisées par une forme rebondie - dite cuboïdale - ainsi que par une forte expression d’enzymes (telles que la fucosyltransférase-VII [FucTVII], la L-selectin ligand sulfotransferase [LSST]) dédiées à la synthèse de molécules indispensables à la traversée vasculaire des lymphocytes (telles que les sialomucines membranaires hypersulfatées décorées par l’antigène reconnu par l’anticorps spécifique MECA-79) ». Extrait de MedSci (Paris) 2012 ; 28 : 347–9.
Les chimiokines possèdent quatre résidus cystéine conservés, et sont classées en deux sous-groupes selon la position de deux de quatre résidus cystéine : chimiokines C-C : les cystéines conservées sont contiguës ; chimiokines C-X-C : les cystéines conservées sont séparées par d’autres acides aminés.
La maladie de Castelman (MC) se caractérise par une hypertrophie ganglionnaire avec hyperplasie lymphoïde angiofolliculaire. Il existe une forme localisée (la plus fréquente), limitée à un site ganglionnaire, et une forme multicentrique (rare), où plusieurs sites sont touchés. L’étiologie de la MC est encore mal connue mais plusieurs études confirment le rôle du virus HHV8, par ailleurs responsable du sarcome de Kaposi (Orphanet, 2006).
Références
- Galie N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2009 ; 34 : 1219–1263. [CrossRef] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004 ; 351 : 1425–1436. [CrossRef] [PubMed] [Google Scholar]
- Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009 ; 54 : S43–S54. [CrossRef] [PubMed] [Google Scholar]
- Girerd B, Montani D, Eyries M, et al. Absence of influence of gender, BMPR2 mutation type on clinical phenotypes of pulmonary arterial hypertension. Respir Res 2010 ; 11 : 73. [CrossRef] [PubMed] [Google Scholar]
- O’Callaghan DS, Savale L, Montani D, et al. Treatment of pulmonary arterial hypertension with targeted therapies. Nat Rev Cardiol 2011 ; 8 : 526–538. [CrossRef] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010 ; 122 : 156–163. [CrossRef] [MathSciNet] [PubMed] [Google Scholar]
- Perros F, Montani D, Dorfmuller P, et al. Nouvelles approches immunopathologiques de l’HTAP. Presse Med 2011 ; 40 Suppl 1 : 1S3–13. [CrossRef] [PubMed] [Google Scholar]
- Tamby MC, Chanseaud Y, Humbert M, et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax 2005 ; 60 : 765–772. [CrossRef] [PubMed] [Google Scholar]
- Terrier B, Tamby MC, Camoin L, et al. Antifibroblast antibodies from systemic sclerosis patients bind to {alpha}-enolase and are associated with interstitial lung disease. Ann Rheum Dis 2010 ; 69 : 428–433. [CrossRef] [PubMed] [Google Scholar]
- Terrier B, Tamby MC, Camoin L, et al. Identification of target antigens of antifibroblast antibodies in pulmonary arterial hypertension. Am J Respir Crit Care Med 2008 ; 177 : 1128–1134. [CrossRef] [PubMed] [Google Scholar]
- Jais X, Launay D, Yaici A, et al. Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: a retrospective analysis of twenty-three cases. Arthritis Rheum 2008 ; 58 : 521–531. [CrossRef] [PubMed] [Google Scholar]
- Pugh ME, Hemnes AR. Development of pulmonary arterial hypertension in women: interplay of sex hormones and pulmonary vascular disease. Womens Health (Lond Engl) 2010 ; 6 : 285–296. [CrossRef] [PubMed] [Google Scholar]
- Quintero OL, Amador-Patarroyo MJ, Montoya-Ortiz G, et al. Autoimmune disease and gender: plausible mechanisms for the female predominance of autoimmunity. J Autoimmun 2012 ; 38 : J109–J119. [CrossRef] [PubMed] [Google Scholar]
- Larkin EK, Newman JH, Austin ED, et al. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 2012 ; 186 : 892–896. [CrossRef] [PubMed] [Google Scholar]
- Tofovic SP. Estrogens and development of pulmonary hypertension: interaction of estradiol metabolism and pulmonary vascular disease. J Cardiovasc Pharmacol 2010 ; 56 : 696–708. [CrossRef] [PubMed] [Google Scholar]
- Nicolls MR, Taraseviciene-Stewart L, Rai PR, et al. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J 2005 ; 26 : 1110–1118. [CrossRef] [PubMed] [Google Scholar]
- Perros F, Dorfmuller P, Montani D, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2012 ; 185 : 311–321. [CrossRef] [PubMed] [Google Scholar]
- Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a french nationwide prospective multicenter study. Arthritis Rheum 2005 ; 52 : 3792–3800. [CrossRef] [PubMed] [Google Scholar]
- Fois E, Le Guern V, Dupuy A, et al. Noninvasive assessment of systolic pulmonary artery pressure in systemic lupus erythematosus: retrospective analysis of 93 patients. Clin Exp Rheumatol 2010 ; 28 : 836–841. [PubMed] [Google Scholar]
- Chu JW, Kao PN, Faul JL, et al. High prevalence of autoimmune thyroid disease in pulmonary arterial hypertension. Chest 2002 ; 122 : 1668–1673. [CrossRef] [PubMed] [Google Scholar]
- Sitbon O, Lascoux-Combe C, Delfraissy JF, et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J Respir Crit Care Med 2008 ; 177 : 108–113. [CrossRef] [PubMed] [Google Scholar]
- Lapa M, Dias B, Jardim C, et al. Cardiopulmonary manifestations of hepatosplenic schistosomiasis. Circulation 2009 ; 119 : 1518–1523. [CrossRef] [PubMed] [Google Scholar]
- Austin ED, Rock MT, Mosse CA, et al. T lymphocyte subset abnormalities in the blood and lung in pulmonary arterial hypertension. Respir Med 2010 ; 104 : 454–462. [CrossRef] [PubMed] [Google Scholar]
- Ulrich S, Nicolls MR, Taraseviciene L, et al. Increased regulatory and decreased CD8+ cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration 2008 ; 75 : 272–280. [CrossRef] [PubMed] [Google Scholar]
- Huertas A, Tu L, Gambaryan N, et al. Leptin and regulatory T-lymphocytes in idiopathic pulmonary arterial hypertension. Eur Respir J 2012 ; 40 : 895–904. [CrossRef] [PubMed] [Google Scholar]
- Savai R, Pullamsetti SS, Kolbe J, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2012 ; 186 : 897–908. [CrossRef] [PubMed] [Google Scholar]
- Tamosiuniene R, Tian W, Dhillon G, et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 2011 ; 109 : 867–879. [CrossRef] [PubMed] [Google Scholar]
- Perros F, Cohen-Kaminsky S, Humbert M. Understanding the role of CD4+CD25(high) (so-called regulatory) T cells in idiopathic pulmonary arterial hypertension. Respiration 2008 ; 75 : 253–256. [CrossRef] [PubMed] [Google Scholar]
- Karmochkine M, Cacoub P, Dorent R, et al. High prevalence of antiphospholipid antibodies in precapillary pulmonary hypertension. J Rheumatol 1996 ; 23 : 286–290. [PubMed] [Google Scholar]
- Riboldi P, Gerosa M, Raschi E, et al. Endothelium as a target for antiphospholipid antibodies. Immunobiology 2003 ; 207 : 29–36. [CrossRef] [PubMed] [Google Scholar]
- Bussone G, Tamby MC, Calzas C, et al. IgG from patients with pulmonary arterial hypertension and/or systemic sclerosis binds to vascular smooth muscle cells and induces cell contraction. Ann Rheum Dis 2012 ; 71 : 596–605. [CrossRef] [PubMed] [Google Scholar]
- Renaudineau Y, Dugue C, Dueymes M, et al. Antiendothelial cell antibodies in systemic lupus erythematosus. Autoimmun Rev 2002 ; 1 : 365–372. [CrossRef] [PubMed] [Google Scholar]
- Negi VS, Tripathy NK, Misra R, et al. Antiendothelial cell antibodies in scleroderma correlate with severe digital ischemia and pulmonary arterial hypertension. J Rheumatol 1998 ; 25 : 462–466. [PubMed] [Google Scholar]
- Humbert M, Monti G, Brenot F, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 1995 ; 151 : 1628–1631. [CrossRef] [PubMed] [Google Scholar]
- Farha S, Sharp J, Asosingh K, et al. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm Circ 2012 ; 2 : 220–228. [CrossRef] [PubMed] [Google Scholar]
- Benoist C, Mathis D. Mast cells in autoimmune disease. Nature 2002 ; 420 : 875–878. [CrossRef] [PubMed] [Google Scholar]
- Arends SJ, Damoiseaux J, Duijvestijn A, et al. Prevalence of anti-endothelial cell antibodies in idiopathic pulmonary arterial hypertension. Eur Respir J 2010 ; 35 : 923–925. [CrossRef] [PubMed] [Google Scholar]
- Rose NR. Bona C Defining criteria for autoimmune diseases (Witebsky’s postulates revisited). Immunol Today 1993 ; 14 : 426–430. [CrossRef] [PubMed] [Google Scholar]
- Yeager M, Cripe PJ, Colvin KL, et al. Loss of tolerance associated with bronchus associated lymphoid tissue expansion in pulmonary hypertension. Am J Respir Crit Care Med 2012 ; 185 : A4761. [Google Scholar]
- Quismorio FP, Jr, Sharma O, Koss M, et al. Immunopathologic and clinical studies in pulmonary hypertension associated with systemic lupus erythematosus. Semin Arthritis Rheum 1984 ; 13 : 349–359. [CrossRef] [PubMed] [Google Scholar]
- Carragher DM, Rangel-Moreno J, Randall TD. Ectopic lymphoid tissues and local immunity. Semin Immun 2008 ; 20 : 26–42. [CrossRef] [Google Scholar]
- Price LC, Montani D, Tcherakian C, et al. Dexamethasone reverses monocrotaline-induced pulmonary arterial hypertension in rats. Eur Respir J 2011 ; 37 : 813–822. [CrossRef] [PubMed] [Google Scholar]
- Ogawa A, Nakamura K, Mizoguchi H, et al. Prednisolone ameliorates idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2011 ; 183 : 139–140. [CrossRef] [PubMed] [Google Scholar]
- Hennigan S, Channick RN, Silverman GJ. Rituximab treatment of pulmonary arterial hypertension associated with systemic lupus erythematosus: a case report. Lupus 2008 ; 17 : 754–756. [CrossRef] [PubMed] [Google Scholar]
- Semerano L, Boissier MC. Les anticorps monoclonaux dans les maladies immunes inflammatoires chroniques. Med Sci (Paris) 2009 ; 25 : 1108–1112. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Tanaka Y. Diagnosis and therapy for systemic lupus erythematosus. Nihon Naika Gakkai Zasshi 2007 ; 96 : 2159–2164. [CrossRef] [PubMed] [Google Scholar]
- Sfikakis PP, Boletis JN, Lionaki S, et al. Remission of proliferative lupus nephritis following B cell depletion therapy is preceded by down-regulation of the T cell costimulatory molecule CD40 ligand: an open-label trial. Arthritis Rheum 2005 ; 52 : 501–513. [CrossRef] [PubMed] [Google Scholar]
- Rincon M. Interleukin-6: from an inflammatory marker to a target for inflammatory diseases. Trends Immunol 2012 ; 33 : 571–577. [CrossRef] [PubMed] [Google Scholar]
- Kishimoto T. Interleukin-6 and its receptor in autoimmunity. J Autoimmun 1992 ; 5 Suppl A : 123–132. [CrossRef] [PubMed] [Google Scholar]
- Steiner MK, Syrkina OL, Kolliputi N, et al. Interleukin-6 overexpression induces pulmonary hypertension. Circulation Res 2009 ; 104 : 236–244. [Google Scholar]
- Soon E, Holmes AM, Treacy CM, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010 ; 122 : 920–927. [CrossRef] [PubMed] [Google Scholar]
- Karavolias GK, Georgiadou P, Gkouziouta A, et al. Short and long term anti-inflammatory effects of bosentan therapy in patients with pulmonary arterial hypertension: relation to clinical and hemodynamic responses. Expert Opin Ther Targets 2010 ; 14 : 1283–1289. [CrossRef] [PubMed] [Google Scholar]
- Furuya Y, Satoh T, Kuwana M., Interleukin-6 as a potential therapeutic target for pulmonary arterial hypertension. Int J Rheumatol 2010 ; 2010 : 720305. [PubMed] [Google Scholar]
- Arita Y, Sakata Y, Sudo T, et al. The efficacy of tocilizumab in a patient with pulmonary arterial hypertension associated with Castleman’s disease. Heart Vessels 2010 ; 25 : 444–447. [CrossRef] [PubMed] [Google Scholar]
- Taniguchi K, Shimazaki C, Fujimoto Y, et al. Tocilizumab is effective for pulmonary hypertension associated with multicentric Castleman’s disease. Int J Hematol 2009 ; 90 : 99–102. [CrossRef] [PubMed] [Google Scholar]
- Terrier B, Mouthon L. Lupus érythémateux systémique. Med Sci (Paris) 2013 ; 29 : 65–73. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Cartron G, Rossi JF. Anticorps monoclonaux thérapeutiques en oncohématologie. Med Sci (Paris) 2009 ; 25 : 1085–1089. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Néogenèse lymphoïde dans les lésions vasculaires pulmonaires de patients avec HTAP idiopathique. Nous émettons l’hypothèse que les auto-anticorps dirigés contre les fibroblastes et les cellules endothéliales fréquemment trouvés dans le sérum des patients émergent suite à la sélection positive et à l’activation de cellules B autoréactives au sein des follicules lymphoïdes tertiaires. Ces cellules B se développent dans l’adventice des petites artères pulmonaires remodelées dans le poumon des patients avec HTAP, fournissant ainsi un microenvironnement optimal pour la génération d’auto-anticorps pathogènes. À l’image des follicules lymphoïdes secondaires trouvés dans les ganglions, ces follicules lymphoïdes tertiaires sont organisés en zones T et B distinctes avec des centres germinatifs Bcl2-Ki-67+. Des antigènes du soi dérivés de composants vasculaires pourraient atteindre ces follicules lymphoïdes tertiaires par les conduits ER-TR7+ formés par des fibroblastes réticulaires et par des vaisseaux lymphatiques périvasculaires dilatés, et pourraient être présentés aux cellules T et B autoréactives par des cellules dendritiques myéloïdes. Les cellules dendritiques folliculaires exposent les complexes immuns à leur surface et activent les lymphocytes B. Les cellules inflammatoires constituant les follicules lymphoïdes tertiaires pourraient provenir des vasa vasorum se développant dans l’adventice des artères pulmonaires remodelées. Les cellules T naïves, pourraient également entrer dans les structures lymphoïdes tertiaires via des HEV (high endothelial venules) très différenciées, et être polarisées vers le phénotype Th17 favorable à l’induction de l’autoimmunité. De plus les cellules T helper folliculaires pourraient apporter l’aide nécessaire aux cellules B des centres germinatifs (centrocytes). Il en résulterait des hypermutations somatiques, des recombinaisons pour la commutation de classe et la sélection de cellules B de haute affinité. Les vaisseaux lymphatiques afférents dilatés et le réseau de vasa vasorum pourraient apporter aux structures lymphoïdes tertiaires les (auto)antigènes pulmonaires, les cellules dendritiques chargées en (auto)antigènes et les cellules T mémoires (extrait de [17], avec l’autorisation de © Am J Respir Crit Care Med). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.