")
")
| Issue |
Med Sci (Paris)
Volume 28, Number 12, Décembre 2012
|
|
|---|---|---|
| Page(s) | 1095 - 1101 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/20122812020 | |
| Published online | 21 décembre 2012 | |
Génétique des populations et immunité chez l’homme
Le cas des interférons
Population genetics and human immunity: the interferon paradigm
1
Institut Pasteur, département génomes et génétique, unité génétique évolutive humaine, 25, rue du Docteur Roux, 75015 Paris, France
2
CNRS URA3012, 75015 Paris, France
3
Université Pierre et Marie Curie (UPMC), cellule Pasteur, 25, rue du Docteur Roux, 75015 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La génétique évolutive de l’infection, discipline complémentaire aux études cliniques et épidémiologiques, étudie les effets de la sélection naturelle sur notre génome et, plus particulièrement, sur les gènes liés à l’immunité. Les interférons (IFN) sont des cytokines qui jouent un rôle clé dans la réponse de l’hôte humain aux pathogènes. Malgré les nombreuses études effectuées sur ces molécules, la contribution relative des différents IFN à la survie de l’homme reste largement méconnue. Des travaux récents, basés sur une approche de génétique évolutive, ont permis de mettre à jour que les IFN différaient dans leur importance biologique : de redondants, comme IFNA10, à essentiels, comme IFNA8 ou IFNG. De plus, certains polymorphismes au sein des IFN de type III, qui permettent vraisemblablement une plus grande protection contre les infections virales, ont été sélectionnés dans les populations d’origine européenne et asiatique.
Abstract
The evolutionary genetics dissection of immunity-related genes provides insights into immunological defence mechanisms and highlight host pathways playing an important role in pathogen resistance. Recent population genetic data have increased knowledge of the biological relevance of human interferons (IFN), cytokines released by host cells in response to pathogen presence or tumour cells. Some IFN-α subtypes as well as IFN-γ are strongly evolutionarily constrained, suggesting that the functions they fulfil are essential and non redundant. Other IFN, the most extreme cases being IFN-α10 and IFN-ε, can accumulate missense or nonsense mutations at high population frequencies, suggesting higher redundancy. Furthermore, genetic variation at some IFN genes can be advantageous for the host and increase in frequency by positive selection. This has been shown for type III IFN, where mutations at IL28A, IL28B and IL29 have been positively selected in Europeans and Asians, most likely by increasing resistance to viral infection. This review uses the IFN paradigm to illustrate the value of the evolutionary approach in highlighting important determinants of host immune responsiveness in the natural setting.
© 2012 médecine/sciences – Inserm / SRMS
La diversité génétique de l’hôte humain
L’étude de la diversité génétique humaine est d’une importance capitale pour identifier les bases génétiques de notre variabilité phénotypique, en particulier face aux maladies. L’arrivée de nouvelles technologies à haut débit, comme les puces de génotypage « génome entier » ou le séquençage de nouvelle génération, a permis la comparaison des diversités des populations humaines au niveau génomique. Le Human Genome Project [1], l’International Hapmap Project [2] et, plus récemment, le 1000 Genomes Project [3] ont abouti à la découverte de plus de 15 millions de variants génétiques. La plupart sont des single nucleotide polymorphisms (SNP), qui constituent un élément clé pour mesurer l’étendue de la variabilité génétique populationnelle que l’on trouve dans notre espèce, et mieux comprendre ainsi la relation entre variabilité génétique et vulnérabilité aux maladies. Dans ce cadre, le 1000 genomes project a estimé que chaque individu porte entre 10 000 et 11 000 mutations non synonymes qui modifient la séquence en acides aminés, la plupart n’ayant vraisemblablement pas de conséquence phénotypique [3]. Cependant, il a été également estimé que chaque individu possède 250 à 300 gènes ayant des mutations conduisant à une perte potentielle de fonction, ainsi que 50 à 100 mutations (à l’état hétérozygote) déjà associées à des maladies génétiques [3].
La dérive génétique (variation aléatoire des fréquences alléliques au sein d’une population au cours des générations) et l’histoire démographique des populations humaines (taille de la population et migration) ont façonné la diversité génétique, ayant ou non un impact sur le phénotype que l’on retrouve actuellement au niveau de la population. En outre, la sélection naturelle, qui représente l’adaptation génétique d’une population à son environnement, qu’il soit climatique, pathogénique ou nutritionnel, a également eu un impact majeur sur la diversité de notre génome [4]. Par exemple, la fréquence d’une mutation conférant une meilleure résistance à une maladie infectieuse donnée, comme l’allèle de l’hémoglobine S (HbS) qui protège contre le paludisme à Plasmodium falciparum, peut augmenter dans la population par sélection positive. La sélection naturelle a donc une influence majeure sur notre diversité, qu’elle soit génétique ou phénotypique.
De la génétique évolutive à l’immunologie
L’homme, comme tout être vivant, a été tout au long de son histoire, et reste encore aujourd’hui, confronté quotidiennement aux agressions de pathogènes. Nous sommes les descendants de ceux qui ont survécu à l’ensemble des pathogènes auxquels ils ont été exposés. En effet, les maladies infectieuses représentent une pression de sélection majeure et seuls les individus les plus résistants survivent et peuvent se reproduire. Ceci était encore vrai jusqu’à la fin du xixe siècle avant le contrôle des infections par l’hygiène, les antibiotiques et les vaccins, qui a largement augmenté l’espérance de vie dans les pays où l’accès aux soins est aisé [5]. Ainsi, la génétique évolutive de l’infection, discipline complémentaire des études cliniques et épidémiologiques, étudie les effets de la sélection naturelle sur notre génome et, plus particulièrement, sur les gènes liés à l’immunité ou aux interactions hôte-pathogènes [6, 7]. Ces études nous permettent de mieux comprendre la pertinence biologique des différents acteurs de la réponse immunitaire, et de distinguer parmi les gènes ceux ayant joué un rôle essentiel dans notre survie face aux agents infectieux de ceux qui auraient un rôle plus redondant dans nos défenses immunitaires. À l’échelle du génome entier, différentes études ont montré que les gènes de l’immunité, ou plus généralement ceux intervenant dans les interactions hôte-pathogènes, ont été des cibles préférentielles de la sélection naturelle [6, 8, 9]. Nous avons identifié, par exemple, quelques 360 gènes ayant un rôle dans l’immunité et montrant des signatures de sélection positive récente (inférieure à 30 000 ans) [6]. Ces résultats suggèrent que notre système immunitaire a été particulièrement mis à l’épreuve durant les phases les plus récentes de notre évolution, ce qui peut être expliqué par l’explosion des maladies infectieuses au début du Néolithique, il y a 10 000 ans, avec la sédentarisation et le développement de l’agriculture.
Dans le cadre de la génétique évolutive de l’infection, nos études se sont essentiellement concentrées sur les effets de la sélection naturelle sur des familles de gènes codant des récepteurs impliqués dans la réponse immunitaire innée, notamment les pattern recognition receptors (PRR) [10]. En effet, l’immunité innée constitue la première ligne de défense et d’interaction entre l’hôte et les pathogènes : les cellules effectrices de l’immunité innée arborent des PRR qui reconnaissent des motifs moléculaires conservés et partagés par de larges groupes de microbes [10]. Nous avons, par exemple, étudié la diversité génétique au niveau de la population des 10 membres de la famille des récepteurs Toll-like (TLR) [11]. Les TLR sont des senseurs antimicrobiens majeurs répondant à une grande variété de motifs pathogéniques (virus, bactéries et autres). Nous avons pu montrer que les TLR endosomiques (TLR3, TLR7, TLR8 et TLR9), principalement impliqués dans la reconnaissance d’acides nucléiques exogènes, étaient sous des contraintes sélectives extrêmes (sélection purificatrice), suggérant leur rôle primordial et non redondant dans notre système immunitaire. En revanche, les TLR exprimés à la surface cellulaire montrent un relâchement de la contrainte sélective, accumulant ainsi des mutations non synonymes et STOP à des fréquences élevées dans la population, ce qui souligne une redondance immunologique plus importante [11].
Les interférons chez l’homme : ce que l’on sait et ce que l’on ne sait pas
En aval de la reconnaissance des pathogènes par les PRR, les interférons (IFN) sont parmi les premières molécules à être produites. Découverts il y a plus de 50 ans, les IFN sont surtout connus pour leur activité antivirale [12], mais sont également capables de moduler de nombreuses fonctions biologiques majeures, telles que la prolifération, la différenciation et l’activation de plusieurs types cellulaires comme les cellules T, les cellules natural killer, les monocytes, les macrophages et les cellules dendritiques [13]. Les IFN humains ont été classés en trois groupes selon les récepteurs qu’ils utilisent, leur homologie et leur localisation chromosomique (Figure 1) [13]. Les IFN de type I sont au nombre de 17 (13 sous-types d’IFN-α, et les IFN-β/ε/κ/ω ; les gènes codant ces IFN sont localisés sur le chromosome 9) et se lient à un même récepteur composé des sous-unités IFNAR1 et IFNAR2 [14]. L’IFN-γ, le seul IFN de type II, est le produit d’un gène localisé sur le chromosome 12, et utilise un récepteur composé des sous-unités IFN-γR1 et IFN-γR2 [13]. Décrits plus récemment, les IFN de type III constituent un groupe de trois cytokines : IL-28A, IL-28B et IL-29 (aussi connues sous les noms respectifs d’IFN-λ2, IFN-λ3 et IFN-λ1) dont les gènes sont localisés sur le chromosome 19 [15]. Les IFN de type III activent une voie de signalisation similaire à celle des IFN de type I, mais ils utilisent un récepteur différent composé des sous-unités IL-28RA et IL10-RB [16]. Les IFN de type I et III sont très différents de l’IFN-γ (type II). En effet, alors que les IFN de type I et III possèdent un pouvoir antiviral important, l’IFN-γ exerce une plus grande activité antibactérienne, antiparasitaire et antifongique (Figure 2) [13, 17].
 |
Figure 1. Structure génomique des différentes familles d’IFN. Les gènes codant les 17 IFN de type I, dépourvus d’introns, sont localisés dans une région d’environ 400 Kb du chromosome 9, à l’exception de l’IFNK qui est localisé à 6 Mb de cette région. Le gène codant l’IFN de type II, ou IFN-γ, est situé sur le chromosome 12, et les trois gènes codant les IFN de type III, IL28A, IL28B et IL29, sont localisés dans une région d’environ 50 Kb du chromosome 19. |
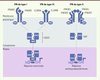 |
Figure 2. Voies de signalisation simplifiées des IFN. Les IFN de type I (IFN-α/β/ε/κ/ω) et de type III (IFN-λ) présentent des voies de signalisation très similaires et aboutissent à l’activation de nombreuses cibles impliquées dans la défense de l’hôte humain contre les pathogènes viraux. La voie de signalisation de l’IFN de type II (IFN-γ) aboutit à une activation de l’immunité antimycobactérienne. JAK1 : Janus kinase 1 ; IRF9 : interferon regulatory factor 9 ; ISGF3 : interferon-stimulated gene factor 3 ; ISRE : interferon stimulation response element ; GAF : IFN-γ-activated factor ; GAS : IFN-γ-activated site. |
Des études de génétique humaine ont permis l’identification de plusieurs variants affectant la production ou la réponse aux IFN à la fois dans des maladies mendéliennes et des maladies complexes, ce qui a permis de mettre à jour les véritables fonctions des IFN in natura [17]. En effet, des mutations au sein de la voie de l’IFN-γ, par exemple dans IFNGR1 et IFNGR2, confèrent une prédisposition mendélienne aux maladies mycobactériennes [18], tandis que des mutations dans les voies des IFN de type I ou III entraîneront de fortes prédispositions aux infections virales [19]. Plusieurs études de génétique épidémiologique ont pu mettre en évidence que des polymorphismes dans la région chromosomique des IFN de type III étaient associés à une clairance spontanée du virus de l’hépatite C (VHC) et à la réponse au traitement contre le VHC [20–23].
Malgré toutes ces avancées majeures, de nombreuses questions restent toujours sans réponse. Les différents membres d’une famille d’IFN possèdent-ils la même importance biologique étant donné le grand nombre d’IFN se liant à un même récepteur (cas des IFN de type I et de type III) ? Certains IFN sont-ils essentiels pour combattre les infections, alors que d’autres présentent une redondance immunologique plus importante ? L’IFN-γ, qui n’est pas une cytokine antivirale, présente-t-il une signature évolutive différente ? Des variations génétiques particulières dans les IFN ont-elles conféré un avantage sélectif à l’hôte, associé à une résistance accrue aux maladies infectieuses ?
Une dissection « évolutive » des interférons humains
Nous avons tenté de répondre à ces questions à l’aide de la génétique évolutive qui nous a permis de comprendre dans quelle mesure les pressions de sélection exercées par les infections ont façonné la variabilité des IFN humains [24]. À cet effet, nous avons séquencé les 21 gènes codant les IFN ainsi que les six gènes codant leurs récepteurs respectifs, dans un panel d’individus sains provenant de différentes origines géographiques et ethniques (Afrique sub-Saharienne, Europe et Asie de l’Est). Nous avons ensuite utilisé ces données de séquence pour mesurer, en utilisant différents tests statistiques, l’étendue de la sélection naturelle sur les IFN aussi bien au niveau interespèce (homme versus chimpanzé) qu’entre différentes populations humaines. Cette étude nous a permis de montrer que les différentes familles d’IFN, ainsi que les différents membres de chaque famille, ont suivi des trajectoires évolutives très différentes.
Les IFN de type I : compromis entre caractère essentiel et redondance
En premier lieu, les IFN de type I présentent des différences très importantes en termes de contraintes sélectives. Alors que certains IFN de type I semblent sous sélection purificatrice (IFNA6, IFNA8, IFNA13, IFNA14), d’autres subissent des contraintes plus relâchées par la présence, à fréquences élevées, de polymorphismes non synonymes (IFNA1, IFNA4, IFNA7, IFNA10, IFNA16 et IFNA17) (Figure 3). À l’extrême, IFNA10 et IFNE présentent même des mutations STOP que l’on trouve à l’état homozygote dans la population générale, suggérant que ces gènes sont peut-être en train de subir une pseudogénisation. Nous avons également souligné que certains polymorphismes non synonymes, la plupart de faible fréquence, avaient pu être introduits par un processus de conversion génique (transfert non réciproque d’une séquence d’ADN entre deux brins d’ADN ayant une grande homologie), étant donné que ces gènes sont des paralogues encore très proches génétiquement. Cette observation suggère qu’au-delà de la duplication, la conversion génique a contribué à l’évolution des IFN de type I chez les mammifères [25].
 |
Figure 3. Variabilité fonctionnelle des membres des différentes familles d’IFN. Proportion de chromosomes porteurs d’au moins un variant non synonyme ou STOP dans la population générale. La portion rouge des camemberts correspond à la proportion de chromosomes porteurs d’au moins un polymorphisme non synonyme, la portion noire à la proportion de chromosomes porteurs d’au moins un polymorphisme STOP, et la portion bleue à la proportion de chromosomes ne portant ni variant non synonyme ni STOP. Les gènes inclus dans des rectangles gris correspondent aux gènes codant les sous-unités des récepteurs des IFN. |
Nos découvertes fournissent une preuve de la complexité des actions biologiques des IFN de type I. Chez l’homme, des immunodéficiences primaires de la voie des IFN de type I, incluant des déficiences de STAT-1 (signal transducer and activator of transcription-1) et TYK-2 (tyrosine kinase 2), ont montré que les IFN de type I sont très importants dans l’immunité antivirale [17]. L’intégration de nos données de génétique des populations dans un cadre clinique nous indique donc qu’au moins un sous-groupe d’IFN de type I joue un rôle essentiel et non redondant dans l’immunité antivirale et dans des conditions naturelles. Le fait de trouver d’autres sous-types accumulant des mutations non synonymes ou STOP suggère que ceux-ci remplissent des fonctions déjà occupées par d’autres sous-types. L’existence d’un si grand nombre de sous-types d’IFN de type I et les différences dans leurs degrés de diversité et de redondance peuvent souligner la grande capacité d’évolution du système de défense de l’hôte dans le but de développer des réponses antivirales optimales.
Cependant, il a également été montré que l’activité des IFN de type I pouvait être néfaste pour l’hôte aussi bien dans un contexte infectieux qu’auto-immun [26]. De plus, il semblerait que ces IFN puissent avoir des rôles opposés dans les infections virales et bactériennes [26]. Un tel mécanisme est illustré par IFN-α8 et IFN-α13, qui sont tous les deux sous de très fortes contraintes sélectives. Pourtant, ils présentent un pouvoir antiviral très différent : très fort pour IFN-α8 et beaucoup plus faible pour IFN-α13 [27–29]. Cependant, les différences de bioactivité entre les sous-types d’IFN de type I ne vont pas seulement dépendre de leur pouvoir antiviral respectif et de leur affinité pour leur récepteur, mais également de leur production. Peu d’études ont mesuré de manière systématique le niveau d’expression de chaque sous-type d’IFN de type I [30, 31], et on ignore lesquels sont les plus exprimés au sein des différentes populations cellulaires. En regard de ces observations, nous pouvons émettre l’hypothèse que le maintien et la sélection de pouvoirs antiviraux et/ou d’une production cellulaire variée sont dus à la régulation de l’activité globale des IFN de type I.
L’IFN de type II ou IFN-γ : seul et unique !
IFNG, codant le seul IFN de type II, est le seul gène parmi ceux codant les trois familles d’IFN et leurs récepteurs à ne présenter aucun polymorphisme modifiant la séquence en acides aminés [24, 32]. Ce gène est sujet à la sélection purificatrice la plus extrême au sein des IFN. IFNG se trouve même parmi les 10 % des gènes liés à l’immunité qui subissent les contraintes sélectives les plus intenses sur les variations non synonymes dans le génome humain [32]. Des études de génétique clinique ont démontré que six gènes impliqués dans la voie de l’IFN-γ (IL-12/23–IFN-γ) jouent un rôle clé dans l’immunité [18]. En particulier, des troubles de la production d’IFN-γ (causés par des mutations affectant IL-12B, IL-12RB1 ou NEMO, NF-κB essential modulator) et de la réponse à l’IFN-γ (causés par des mutations touchant IFNGR1, IFNGR2 ou STAT1) sont associés à une susceptibilité mendélienne aux maladies mycobactériennes chez des patients pourtant résistants à la plupart des virus [17]. En effet, nos données de génétique des populations indiquent que, même si les gènes codant les deux sous-unités du récepteur de l’IFN-γ montrent un relâchement de la contrainte sélective, tout particulièrement IFNGR2, ces deux gènes tolèrent très peu de changements qui pourraient avoir un impact fonctionnel majeur (mutations non synonymes). L’intégration de ces données avec celles de la génétique clinique montre que les variations génétiques ayant un impact significatif sur la fonction protéique des locus impliqués dans la voie de l’IFN-γ ne sont pas tolérées dans la population générale. Ce résultat prouve que cette voie est essentielle à la survie de l’hôte humain et non redondante, y compris dans le cadre de la défense de l’hôte face aux infections mycobactériennes.
Les IFN de type III : mutations avantageuses en Europe et en Asie
Enfin, nous avons également montré que les IFN de type III sont le seul groupe d’IFN qui présente des signatures de sélection positive spécifiques à certaines populations humaines [24]. Nous avons identifié huit mutations (dont quatre correspondent à des mutations non synonymes) dans la région génomique qui englobe les trois IFN de type III (IL28B, IL28A et IL29) qui seraient sous très forte pression de sélection positive en Asie, et également en Europe concernant IL29 (Figure 4). Ceci suggère que ces mutations ont conféré un avantage évolutif pour la survie de l’homme dans ces régions géographiques. On sait aujourd’hui, grâce à des études cliniques, que les IFN de type III ont un rôle majeur dans l’immunité antivirale [17] et que, par conséquent, les pressions agissant sur ces IFN sont vraisemblablement d’origine virale. En effet, les cinq polymorphismes dans la région du gène codant IL28B que nous avons identifiés comme étant les cibles de la sélection positive en Asie (Figure 4), ont été associés à la clairance spontanée du VHC et à une meilleure réponse au traitement associant IFN-α pégylé et Ribavirine dans le cas d’infections chroniques par le VHC, et ce au sein de populations africaines, européennes et asiatiques [20–23]. De manière intéressante, selon les odds ratio (mesure statistique de l’effet relatif d’un allèle), pour les allèles protecteurs, il a été suggéré que des variations au sein de l’IL28B pourraient conférer un effet protecteur plus important chez les populations asiatiques que chez les individus européens et africains [33]. Nos données suggèrent ainsi que, chez les populations asiatiques, les fréquences des allèles protecteurs auraient augmenté sous l’action de la sélection positive la plus forte, plutôt que par simple dérive génétique. Étant donné la nature peu mortelle de la pathogenèse du VHC, il est peu probable que ce virus soit le responsable des pressions de sélection s’exerçant sur IL28B. Il est donc fort possible que d’autres flavivirus plus virulents soient les responsables véritables des signatures de sélection observées.
 |
Figure 4. Variants génétiques sous sélection positive au sein des IFN de type III (IL28B, IL28A et IL29). Les rectangles pleins correspondent aux régions exoniques et les flèches horizontales au-dessus des exons indiquent le sens du cadre de lecture. Les variants génétiques qui affichent des signatures de sélection positive spécifiques de certaines populations sont présentés. Les SNP non synonymes sont représentés en rouge, alors que les SNP localisés dans des régions non codantes sont en noir. Les SNP au sein des gènes IL28A et IL28B sont sous sélection positive dans les populations asiatiques, alors que le SNP au sein du gène IL29 est sous sélection positive à la fois en Europe et en Asie. |
Le fait que les variants au sein de IL28B trouvés ici sous sélection positive soient les mêmes que ceux qui sont associés à la clairance spontanée des infections au VHC montre clairement le pouvoir prédictif de l’approche évolutive, comme complément des études épidémiologiques et de génétique médicale. Ceci est particulièrement important pour les variants positivement sélectionnés au sein des gènes IL28A et IL29 dont l’impact fonctionnel reste à définir. La plus forte signature de sélection observée concerne un polymorphisme non synonyme au sein d’IL29 (Figure 4), en Europe et en Asie. D’autres études sont nécessaires afin de mettre à jour le rôle immunologique des trois IFN de type III - en particulier pour les polymorphismes sous sélection positive - en relation avec la susceptibilité ou la pathogenèse des maladies infectieuses ou auto-immunes.
Conclusion
L’approche de génétique évolutive des populations indique que les membres des trois familles d’IFN diffèrent dans leur importance biologique, allant de très contraints à redondants et non essentiels. L’identification des IFN sujets à de fortes contraintes ou à une sélection positive souligne leur rôle majeur dans l’immunité et ouvre la voie à des études approfondies pour évaluer le potentiel de ces molécules dans le cadre de vaccinations, diagnostics et traitements. Plus généralement, notre étude fournit un paradigme de l’utilisation de la génétique des populations dans le contexte des infections qui devrait accroître notre connaissance et notre compréhension de l’importance biologique des gènes de l’hôte humain liés à l’immunité dans le milieu naturel.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Cette étude a été financée par l’Institut Pasteur, le CNRS, l’Agence nationale de la recherche (ANR-08-MIEN-009-01) et la Fondation pour la recherche médicale (FRM).
Références
- Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature 2001 ; 409 : 860–921. [CrossRef] [PubMed] [Google Scholar]
- Altshuler DM, Gibbs RA, Peltonen L, et al. Integrating common and rare genetic variation in diverse human populations. Nature 2010 ; 467 : 52–58. [CrossRef] [PubMed] [Google Scholar]
- Durbin RM, Abecasis GR, Altshuler DL, et al. A map of human genome variation from population-scale sequencing. Nature 2010 ; 467 : 1061–1073. [CrossRef] [PubMed] [Google Scholar]
- Barreiro LB, Laval G, Quach H, et al. Natural selection has driven population differentiation in modern humans. Nat Genet 2008 ; 40 : 340–345. [CrossRef] [PubMed] [Google Scholar]
- Casanova JL, Abel L. Inborn errors of immunity to infection: the rule rather than the exception. J Exp Med 2005 ; 202 : 197–201. [CrossRef] [PubMed] [Google Scholar]
- Barreiro LB, Quintana-Murci L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet 2010 ; 11 : 17–30. [CrossRef] [PubMed] [Google Scholar]
- Quintana-Murci L, Alcais A, Abel L, Casanova JL. Immunology in natura: clinical, epidemiological and evolutionary genetics of infectious diseases. Nat Immunol 2007 ; 8 : 1165–1171. [CrossRef] [PubMed] [Google Scholar]
- Nielsen R, Hellmann I, Hubisz M, et al. Recent and ongoing selection in the human genome. Nat Rev Genet 2007 ; 8 : 857–868. [CrossRef] [PubMed] [Google Scholar]
- Sabeti PC, Schaffner SF, Fry B, et al. Positive natural selection in the human lineage. Science 2006 ; 312 : 1614–1620. [CrossRef] [PubMed] [Google Scholar]
- Delneste Y, Beauvillain C, Jeannin P. Immunité naturelle : structure et fonction des Toll-like receptors. Med Sci (Paris) 2007 ; 23 : 67–73. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Barreiro LB, Ben-Ali M, Quach H, et al. Evolutionary dynamics of human Toll-like receptors, their different contributions to host defense. PLoS Genet 2009 ; 5 : e1000562. [CrossRef] [PubMed] [Google Scholar]
- Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 1957 ; 147 : 258–267. [CrossRef] [PubMed] [Google Scholar]
- Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev 2004 ; 202 : 8–32. [CrossRef] [PubMed] [Google Scholar]
- Uze G, Schreiber G, Piehler J, Pellegrini S. The receptor of the type I interferon family. Curr Top Microbiol Immunol 2007 ; 316 : 71–95. [CrossRef] [PubMed] [Google Scholar]
- Kotenko SV, Gallagher G, Baurin VV, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 2003 ; 4 : 69–77. [CrossRef] [PubMed] [Google Scholar]
- Kotenko SV, Krause CD, Izotova LS, et al. Identification and functional characterization of a second chain of the interleukin-10 receptor complex. EMBO J 1997 ; 16 : 5894–5903. [CrossRef] [PubMed] [Google Scholar]
- Zhang SY, Boisson-Dupuis S, Chapgier A, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev 2008 ; 226 : 29–40. [CrossRef] [PubMed] [Google Scholar]
- Filipe-Santos O, Bustamante J, Chapgier A, et al. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol 2006 ; 18 : 347–361. [CrossRef] [PubMed] [Google Scholar]
- Chapgier A, Wynn RF, Jouanguy E, et al. Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J Immunol 2006 ; 176 : 5078–5083. [PubMed] [Google Scholar]
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009 ; 461 : 399–401. [CrossRef] [PubMed] [Google Scholar]
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009 ; 461 : 798–801. [CrossRef] [PubMed] [Google Scholar]
- Labie D, Gilgenkrantz H. Variants génétiques du gène IL28B et élimination du virus de l’hépatite C. Med Sci (Paris) 2010 ; 26 : 225–226. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009 ; 41 : 1105–1109. [CrossRef] [PubMed] [Google Scholar]
- Manry J, Laval G, Patin E, et al. Evolutionary genetic dissection of human interferons. J Exp Med 2011 ; 208 : 2747–2759. [CrossRef] [PubMed] [Google Scholar]
- Genin P, Vaccaro A, Civas A. The role of differential expression of human interferon – a genes in antiviral immunity. Cytokine Growth Factor Rev 2009 ; 20 : 283–295. [CrossRef] [PubMed] [Google Scholar]
- Trinchieri G. Type I interferon: friend or foe?. J Exp Med 2010 ; 207 : 2053–2063. [CrossRef] [PubMed] [Google Scholar]
- Foster GR, Rodrigues O, Ghouze F, et al. Different relative activities of human cell-derived interferon-alpha subtypes: IFN-alpha 8 has very high antiviral potency. J Interferon Cytokine Res 1996 ; 16 : 1027–1033. [PubMed] [Google Scholar]
- Jaks E, Gavutis M, Uze G, et al. Differential receptor subunit affinities of type I interferons govern differential signal activation. J Mol Biol 2007 ; 366 : 525–539. [CrossRef] [PubMed] [Google Scholar]
- Lavoie TB, Kalie E, Crisafulli-Cabatu S, et al. Binding and activity of all human alpha interferon subtypes. Cytokine 2011 ; 56 : 282–289. [CrossRef] [PubMed] [Google Scholar]
- Coccia EM, Severa M, Giacomini E, et al. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol 2004 ; 34 : 796–805. [CrossRef] [PubMed] [Google Scholar]
- Genin P, Lin R, Hiscott J, Civas A. Differential regulation of human interferon A gene expression by interferon regulatory factors 3 and 7. Mol Cell Biol 2009 ; 29 : 3435–3450. [CrossRef] [PubMed] [Google Scholar]
- Manry J, Laval G, Patin E, et al. Evolutionary genetics evidence of an essential, nonredundant role of the IFN-gamma pathway in protective immunity. Hum Mutat 2011 ; 32 : 633–642. [CrossRef] [PubMed] [Google Scholar]
- O’Brien TR. Interferon-alfa, interferon-lambda and hepatitis C. Nat Genet 2009 ; 41 : 1048–1050. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1. Structure génomique des différentes familles d’IFN. Les gènes codant les 17 IFN de type I, dépourvus d’introns, sont localisés dans une région d’environ 400 Kb du chromosome 9, à l’exception de l’IFNK qui est localisé à 6 Mb de cette région. Le gène codant l’IFN de type II, ou IFN-γ, est situé sur le chromosome 12, et les trois gènes codant les IFN de type III, IL28A, IL28B et IL29, sont localisés dans une région d’environ 50 Kb du chromosome 19. |
| Dans le texte | |
|
Figure 2. Voies de signalisation simplifiées des IFN. Les IFN de type I (IFN-α/β/ε/κ/ω) et de type III (IFN-λ) présentent des voies de signalisation très similaires et aboutissent à l’activation de nombreuses cibles impliquées dans la défense de l’hôte humain contre les pathogènes viraux. La voie de signalisation de l’IFN de type II (IFN-γ) aboutit à une activation de l’immunité antimycobactérienne. JAK1 : Janus kinase 1 ; IRF9 : interferon regulatory factor 9 ; ISGF3 : interferon-stimulated gene factor 3 ; ISRE : interferon stimulation response element ; GAF : IFN-γ-activated factor ; GAS : IFN-γ-activated site. |
| Dans le texte | |
|
Figure 3. Variabilité fonctionnelle des membres des différentes familles d’IFN. Proportion de chromosomes porteurs d’au moins un variant non synonyme ou STOP dans la population générale. La portion rouge des camemberts correspond à la proportion de chromosomes porteurs d’au moins un polymorphisme non synonyme, la portion noire à la proportion de chromosomes porteurs d’au moins un polymorphisme STOP, et la portion bleue à la proportion de chromosomes ne portant ni variant non synonyme ni STOP. Les gènes inclus dans des rectangles gris correspondent aux gènes codant les sous-unités des récepteurs des IFN. |
| Dans le texte | |
|
Figure 4. Variants génétiques sous sélection positive au sein des IFN de type III (IL28B, IL28A et IL29). Les rectangles pleins correspondent aux régions exoniques et les flèches horizontales au-dessus des exons indiquent le sens du cadre de lecture. Les variants génétiques qui affichent des signatures de sélection positive spécifiques de certaines populations sont présentés. Les SNP non synonymes sont représentés en rouge, alors que les SNP localisés dans des régions non codantes sont en noir. Les SNP au sein des gènes IL28A et IL28B sont sous sélection positive dans les populations asiatiques, alors que le SNP au sein du gène IL29 est sous sélection positive à la fois en Europe et en Asie. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.