")
")
| Issue |
Med Sci (Paris)
Volume 28, Number 8-9, Août–Septembre 2012
|
|
|---|---|---|
| Page(s) | 703 - 706 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2012288010 | |
| Published online | 22 août 2012 | |
KLHL3 et CULLIN-3
Deux nouveaux gènes responsables d’une forme mendélienne d’hypertension artérielle
KLHL3 and CULLIN-3: new genes involved in familial hypertension
1
Inserm, UMRS 970, Centre de recherche PARCC (Paris centre de recherche cardiovasculaire), Paris, France
2
Université Paris Descartes, Paris-Sorbonne-Cité, Faculté de Médecine, Paris, France
3
Assistance publique-hôpitaux de Paris (AP-HP), département de génétique, Hôpital Européen Georges Pompidou (HEGP), 56, rue Leblanc, 75015 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Rôle des gènes WNK1, WNK4 et KLHL3 dans l’hypertension hyperkaliémique familiale
L’étude des formes mendéliennes d’hypertension artérielle, a permis des avancées remarquables dans la compréhension des mécanismes de régulation de la pression artérielle, en dépit de la rareté de ces formes. L’hypertension hyperkaliémique familiale (HHF), aussi connue sous le terme de syndrome de Gordon ou de pseudo-hypoaldostéronisme de type 2, est une de ces formes rares d’hypertension artérielle, transmise essentiellement selon un mode autosomique dominant [1]. Les sujets atteints présentent des anomalies métaboliques associant hyperkaliémie, acidose métabolique et hyperchlorémie en l’absence de toute insuffisance rénale. Ces désordres métaboliques, associés à la grande efficacité des diurétiques thiazidiques révèlent une anomalie primaire de réabsorption ionique au niveau du rein.
En 2001, nous avions identifié, en collaboration avec l’équipe de R.P. Lifton (Université de Yale, New Haven, États-Unis), les deux premiers gènes responsables de cette pathologie [2]. Ces deux gènes, WNK1 et WNK4 (With no K [lysine]), codent pour deux sérine thréonine kinases de structure et d’expression particulièrement complexes [3]. Par la suite, des études in vitro puis in vivo ont montré les fonctions activatrices et inhibitrices de WNK1 et WNK4 sur plusieurs transporteurs ioniques, en particulier le cotransporteur Na+-Cl-, NCC [4].
Cependant, les mutations trouvées dans les gènes WNK1 et WNK4 n’expliquent qu’une partie minoritaire des cas décrits. Un ou plusieurs autres gène(s) devrai(en)t donc être également responsable(s) de cette pathologie. Deux publications récentes viennent de combler cette lacune en identifiant le rôle essentiel d’un mécanisme moléculaire de dégradation protéique par ubiquitination dans la régulation du transport ionique et de la pression artérielle [5, 6]. Les deux équipes - dont la nôtre - ont utilisé les outils modernes de la génétique humaine, en particulier des études d’exome entier, pour la recherche de mutations codantes dans une pathologie rare.
Notre étude a associé deux stratégies : une étude de liaison classique par Lod score1 et la recherche de variants codants par analyse d’exome entier [6]. La recherche d’un intervalle de liaison a été effectuée à partir de deux familles qui ne présentaient pas de mutations dans les gènes WNK1 et WNK4. L’une d’entre elles a été recrutée dans le département de génétique de l’hôpital européen Georges Pompidou (HEGP) à Paris (Dr X. Jeunemaitre), l’autre à l’institut du thorax à Nantes (Inserm UMR 915, Dr J.J. Schott). Une seule région était commune aux deux familles (5q31.2) avec un Lod score cumulé très significatif (9,0). Cette étude de liaison a été complétée par un séquençage d’exome entier chez trois cas atteints et un sujet non atteint de chaque famille. Un seul gène, KLHL3 (Kelch-like 3), présentait, chez les cas atteints des deux familles, des mutations faux-sens considérées comme pathogènes par plusieurs logiciels de prédiction bioinformatique. L’implication du gène KLHL3 a pu être rapidement vérifiée par la coségrégation mutation-pathologie dans les deux familles initiales et par l’identification de 14 autres mutations faux-sens supplémentaires chez 17 autres sujets atteints non apparentés. Dans 13 cas, la mutation était présente à l’état hétérozygote, en accord avec la transmission dominante attendue. Dans quatre cas issus de familles consanguines, la mutation était présente à l’état homozygote, correspondant à une forme apparemment récessive [6]. L’expression forte de KLHL3 dans le néphron et sa colocalisation avec le cotransporteur NCC dans le tubule contourné distal renforçaient les arguments de causalité de ce gène dans la pathologie.
Les mutations de KLHL3 et Cullin-3 modifient l’expression d’un transporteur rénal ionique
À la même période, une équipe américaine identifiait indépendamment par analyse d’exome entier le même gène, KLHL3, comme responsable de la pathologie [5]. Elle répertoria 24 mutations différentes, la plupart faux-sens, mais également non-sens, petites délétions et mutations d’épissage. Seize mutations étaient présentes à l’état hétérozygote, huit à l’état homozygote ou hétérozygote composite, expliquant ainsi la présentation parfois dominante ou récessive de la pathologie. L’ensemble des mutations KLHL3 retrouvées par les deux équipes sont indiquées dans la Figure 1. De plus, l’équipe de R.P. Lifton montrait la présence de mutations hétérozygotes du gène Cullin-3 chez 17 autres sujets atteints. Ces mutations, survenant pour la plupart de novo, étaient détectées dans des formes plus sévères de la pathologie. Toutes correspondaient à des mutations d’épissage de l’exon 9 du gène Cullin-3. Nous avons confirmé par la suite ce même type de mutations chez sept sujets atteints, ayant aussi une forme précoce et sévère de la pathologie.
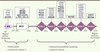 |
Figure 1. Schéma de la protéine KLHL3 et localisation de l’ensemble des mutations retrouvées dans l’hypertension hyperkaliémique familiale. La protéine KLHL3 est composée de 3 domaines différents : un domaine BTB (Bric-à-brac, Tramtrack, Broad complex), responsable de la dimérisation de la protéine ; un domaine BACK (BTB and carboxy-terminal Kelch) et un domaine KELCH, composé de 6 motifs Kelch formant une hélice à 6 pales de feuillets β. Les 37 mutations différentes (22 hétérozygotes et 15 homozygotes représentées en gras) sont essentiellement des mutations faux-sens situées dans le domaine KELCH [5, 6]. |
KLHL3 et culline-3 : partenaires d’un complexe d’ubiquitination de protéines impliquées dans le transport rénal
Que sont donc les protéines Kelch-like 3 et culline-3 ? La protéine Kelch-like 3 appartient à la famille des protéines à domaine BTB-BACK (broad-complex, tramtrack and bric a brac - BTB and C-terminal Kelch) liant l’actine par leur domaine KELCH [7]. Ce domaine est composé de six motifs Kelch (57 acides aminés chacun), séquences permettant de lier des substrats pour la liaison à la culline-3. Les cullines sont des protéines essentielles du complexe E3 ubiquitine ligase dirigeant les protéines cibles vers la dégradation par le protéasome après ubiquitination [8]. La spécificité d’ubiquitination de multiples substrats est obtenue essentiellement par la possibilité d’interaction avec de très nombreuses protéines BTB, dont les protéines Kelch.
Ces éléments orientaient donc vers la présence de mutations « perte de fonction » pour Kelch-like 3 et culline-3, ces deux partenaires participant à un complexe d’ubiquitination de protéines impliquées dans le transport rénal. Le cotransporteur NCC devenait un candidat idéal tant par son rôle connu dans la pathologie que par sa coexpression dans le néphron distal avec Kelch-like 3 [6]. Le niveau d’expression membranaire de NCC est soumis à une régulation complexe, impliquant des mécanismes contrôlant son adressage à la membrane, mais aussi sa dégradation. Une étude récente a mis en évidence que NCC pouvait être soumis à une dégradation dans le réticulum endoplasmique, l’ubiquitination participant à cette dégradation [9]. Un deuxième niveau de régulation, post-traductionnel, de NCC concerne son expression membranaire et fait vraisemblablement intervenir plusieurs mécanismes dont l’endocytose de ce cotransporteur [10].
Kelch-like 3 et culline-3 pourraient participer à ce processus en modulant l’ubiquitination de NCC. Nous avons d’abord testé la régulation conjointe de Kelch-like 3 et NCC dans des conditions hypotoniques, conditions connues comme entraînant une redistribution de NCC des vésicules subapicales cytoplasmiques vers la membrane apicale dans le rein [11]. Sous l’influence d’un milieu hypotonique, l’expression membranaire de NCC dans des cellules HEK293T transfectées augmente de 50 %, alors que celle de Kelch-like 3 est diminuée de 30 %. De plus, l’inhibition de l’expression de Kelch-like 3 par ARN interférence dans un milieu classique augmente l’expression membranaire de NCC, suggérant que KLHL3 est un facteur de régulation négatif de l’expression membranaire de NCC [6].
Modèle d’interaction entre KLHL3-culline-3 et NCC
Un modèle schématique d’interaction entre Kelch-like 3-culline-3 et NCC est résumé dans la Figure 2 . Kelch-like 3 et culline-3 participent à la structure d’un complexe E3 ligase ayant pour cible le cotransporteur NCC dans le néphron distal. En l’absence de mutation, ce complexe permet la dégradation par le protéasome d’une partie des protéines NCC produites dans le réticulum endoplasmique. En présence de mutations des gènes KLHL3 ou Cullin-3, l’interaction des protéines avec leur substrat NCC est altérée et la dégradation du cotransporteur via le protéasome est réduite. En conséquence, un plus grand nombre de molécules NCC seraient présentes à la membrane, entraînant l’augmentation de l’activité de réabsorption rénale d’ions Na+ et Cl-. Cette activité accrue de NCC rend compte de la grande sensibilité des patients aux diurétiques thiazidiques, NCC étant la cible privilégiée de ces agents pharmacologiques.
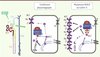 |
Figure 2. Modèle proposé pour expliquer les modifications d’expression du cotransporteur NCC en présence de mutations des gènes KLHL3 et Cullin-3. Un schéma d’un tubule rénal est représenté à gauche. Le cotransporteur Na+-Cl- (NCC) est exprimé dans le tube contourné distal (DCT). Au niveau cellulaire, KLHL3 et culline-3 participent à la régulation de son activité via un processus d’ubiquitination. Culline-3 est une protéine appartenant au complexe E3 ubiquitine ligase dégradant les protéines via le protéasome après ubiquitination. KLHL3 est un partenaire d’interaction de la culline-3, essentiel pour la reconnaissance des substrats, dont NCC. Dans le cas de l’hypertension hyperkaliémique familiale, les mutations de KLHL3 ou culline-3 empêchent l’interaction de NCC avec le complexe E3 ligase, diminuant ainsi sa dégradation, ce qui a pour conséquence une augmentation de son expression et de son activité à la membrane, et donc une absorption accrue de NaCl. CCD : cortical collecting duct ; DCT : distal convoluted tubule ; PCT : proximal convoluted tube ; TAL : thick ascending limb of the loop of Henle. |
Une nouvelle voie de recherche
De nombreux points restent à élucider, parmi lesquels : existe-t-il d’autres transporteurs cibles de ce système d’ubiquitination ? Existe-t-il d’autres protéines Kelch (famille de 38 membres) capables de lier la culline-3 au niveau rénal et de jouer un rôle dans la régulation du transport ionique et de la pression artérielle ? Quelles sont les interactions directes et indirectes entre les différents partenaires ? Quelles sont les relations avec WNK1 et WNK4 dont les mutations sont responsables de la même pathologie ? Comment expliquer que des mutations dans des protéines aussi ubiquitaires entraînent un phénotype essentiellement rénal ?
En conclusion, deux études parallèles ont identifié deux gènes inattendus responsables d’une forme mendélienne d’hypertension artérielle. Les protéines correspondantes appartiennent à un mécanisme de dégradation de molécules impliquées dans le transport ionique. Comme cela fut le cas pour les gènes WNK1 et WNK4 dix ans auparavant, la découverte de mutations dans des gènes codant pour des acteurs insoupçonnés pose de nombreuses questions physiopathologiques et ouvre une nouvelle voie de recherche.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Le Lod score est défini comme le logarithme décimal du rapport de la probabilité d’observation d’une famille sous l’hypothèse d’une liaison d’une distance Q entre deux locus à la probabilité d’observation de cette même famille sous l’hypothèse d’indépendance.
Références
- Gordon RD. Syndrome of hypertension and hyperkalemia with normal glomerular filtration rate. Hypertension 1986 ; 8 : 93–102. [CrossRef] [PubMed] [Google Scholar]
- Wilson FH, Disse-Nicodeme S, Choate KA, et al. Human hypertension caused by mutations in WNK kinase. Science 2001 ; 293 : 1107–1112. [CrossRef] [PubMed] [Google Scholar]
- Hadchouel J, Delaloy C, Jeunemaitre X. WNK1 et WNK4, nouveaux acteurs de l’homéostasie hydrosodée. Med Sci (Paris) 2005 ; 21 : 55–60. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Hadchouel J, Jeunemaitre X. Life and death of the distal nephron: WNK4 and NCC as major players. Cell Metab 2006 ; 4 : 335–337. [CrossRef] [PubMed] [Google Scholar]
- Boyden LM, Choi M, Choate KA, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012 ; 482 : 98–102. [CrossRef] [PubMed] [Google Scholar]
- Louis-Dit-Picard H, Barc J, Trujillano D, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet 2012 ; 44 : 456–460. [CrossRef] [PubMed] [Google Scholar]
- Wilhelm K. BTB protein as henchmen of Cul3-based ubiquitin ligases. Nat Cell Biol 2003 ; 5 : 950–951. [CrossRef] [PubMed] [Google Scholar]
- Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J 2004 ; 23 : 1681–1687. [CrossRef] [PubMed] [Google Scholar]
- Needham PG, Mikoluk K, Dhakarwal P, et al. The thiazide-sensitive NaCl cotransporter is targeted for chaperone-dependent endoplasmic reticulum-associated degradation. J Biol Chem 2011 ; 286 : 43611–43621. [CrossRef] [PubMed] [Google Scholar]
- Mount DB. Regulated endocytosis of NCC. Am J Physiol Renal Physiol 2010 ; 299 : F297–F299. [CrossRef] [PubMed] [Google Scholar]
- Frindt G, Palmer LG. Surface expression of sodium channels and transporters in rat kidney: effects of dietary sodium. Am J Physiol Renal Physiol 2009 ; 297 : F1249–F1255. [CrossRef] [PubMed] [Google Scholar]
© 2012 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1. Schéma de la protéine KLHL3 et localisation de l’ensemble des mutations retrouvées dans l’hypertension hyperkaliémique familiale. La protéine KLHL3 est composée de 3 domaines différents : un domaine BTB (Bric-à-brac, Tramtrack, Broad complex), responsable de la dimérisation de la protéine ; un domaine BACK (BTB and carboxy-terminal Kelch) et un domaine KELCH, composé de 6 motifs Kelch formant une hélice à 6 pales de feuillets β. Les 37 mutations différentes (22 hétérozygotes et 15 homozygotes représentées en gras) sont essentiellement des mutations faux-sens situées dans le domaine KELCH [5, 6]. |
| Dans le texte | |
|
Figure 2. Modèle proposé pour expliquer les modifications d’expression du cotransporteur NCC en présence de mutations des gènes KLHL3 et Cullin-3. Un schéma d’un tubule rénal est représenté à gauche. Le cotransporteur Na+-Cl- (NCC) est exprimé dans le tube contourné distal (DCT). Au niveau cellulaire, KLHL3 et culline-3 participent à la régulation de son activité via un processus d’ubiquitination. Culline-3 est une protéine appartenant au complexe E3 ubiquitine ligase dégradant les protéines via le protéasome après ubiquitination. KLHL3 est un partenaire d’interaction de la culline-3, essentiel pour la reconnaissance des substrats, dont NCC. Dans le cas de l’hypertension hyperkaliémique familiale, les mutations de KLHL3 ou culline-3 empêchent l’interaction de NCC avec le complexe E3 ligase, diminuant ainsi sa dégradation, ce qui a pour conséquence une augmentation de son expression et de son activité à la membrane, et donc une absorption accrue de NaCl. CCD : cortical collecting duct ; DCT : distal convoluted tubule ; PCT : proximal convoluted tube ; TAL : thick ascending limb of the loop of Henle. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.