")
")
| Issue |
Med Sci (Paris)
Volume 28, Number 2, Février 2012
|
|
|---|---|---|
| Page(s) | 200 - 205 | |
| Section | M/S Revues | |
| DOI | https://doi.org/10.1051/medsci/2012282019 | |
| Published online | 27 février 2012 | |
Perturbations de la transcription liées à une dérégulation de P-TEFb : cancer, Sida et hypertrophie cardiaque
Misregulation of P-TEFb activity: pathological consequences
Université de Toulouse, université Paul Sabatier, CNRS laboratoire de biologie moléculaire des eucaryotes, 118, route de Narbonne, F-31062 Toulouse, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Le facteur P-TEFb stimule l’élongation de la transcription par l’ARN pol II. Dans les cellules humaines, le taux de P-TEFb disponible pour activer la transcription est contrôlé par un ARN non codant, l’ARN 7SK. Cet ARN s’associe à une fraction de P-TEFb qu’il séquestre au sein d’un complexe inactif. Un équilibre dynamique entre les formes active et inactive de P-TEFb permet d’adapter sa disponibilité aux besoins transcriptionnels de la cellule. Des perturbations de cet équilibre sont associées au développement de certaines tumeurs et à l’hypertrophie cardiaque. Ce système de régulation est également ciblé par le VIH pour augmenter la fraction active de P-TEFb dans les cellules infectées.
Abstract
P-TEFb stimulates transcription elongation by phosphorylating the carboxy-terminal domain of RNA pol II and antagonizing the effects of negative elongation factors. Its cellular availability is controlled by an abundant non coding RNA, conserved through evolution, the 7SK RNA. Together with the HEXIM proteins, 7SK RNA associates with and sequesters a fraction of cellular P-TEFb into a catalytically inactive complex. Active and inactive forms of P-TEFb are kept in a functional and dynamic equilibrium tightly linked to the transcriptional requirement of the cell. Importantly, cardiac hypertrophy and development of various types of human malignancies have been associated with increased P-TEFb activity, consequence of a disruption of this regulatory equilibrium. In addition, the HIV-1 Tat protein also releases P-TEFb from the 7SK/HEXIM complex during viral infection to promote viral transcription and replication. Here, we review the roles played by the 7SK RNP in cancer development, cardiac hypertrophy and AIDS.
© 2012 médecine/sciences – Inserm / SRMS
Le facteur positif d’élongation de la transcription, P-TEFb, est un hétérodimère composé d’une kinase, CDK9, et d’une cycline (T1 ou T2). L’activité kinase de P-TEFb fut initialement décrite comme essentielle à l’expression des gènes du virus de l’immunodéficience humaine (VIH) et de certains gènes cellulaires (c-myc, c-fos, hsp70). La transcription de ces gènes par l’ARN polymérase (pol) II est principalement contrôlée au cours de l’élongation. En effet, peu après l’initiation de la transcription, l’ARN pol II marque une pause transcriptionnelle, imposée par l’action simultanée de deux facteurs négatifs d’élongation (N-TEF), DSIF (DRB sensitivity inducing factor) et NELF (negative elongation factor) [1, 2]. Le recrutement de P-TEFb au niveau de la machinerie transcriptionnelle permet la phosphorylation par CDK9 du domaine carboxy-terminal (CTD) de l’ARN pol II sur la sérine 2 (Ser2), mais aussi celle des N-TEF (Figure 1). Ces événements de phosphorylation permettent la reprise de la transcription par l’ARN pol II et la synthèse d’ARNm pleine-taille. P-TEFb pourrait être recruté au niveau de la machinerie transcriptionnelle au sein d’un super elongation complex (ou SEC) qui contient un autre facteur d’élongation (la protéine ELL2) et des coactivateurs (les protéines AFF4, ENL et AF9) [3].
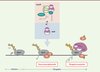 |
Figure 1. Contrôle de l’activité de P-TEFb et élongation de la transcription. P-TEFb existe sous deux formes en équilibre dynamique dans les cellules humaines. Une fraction de P-TEFb est inactivée par l’ARN 7SK et l’autre fraction est sous forme libre et active. La forme active est recrutée au niveau de l’ARN pol II au sein du super elongation complex (SEC) pour lever la pause transcriptionnelle imposée par les facteurs négatifs d’élongation DSIF et NELF. La phosphorylation par P-TEFb du CTD de l’ARN pol II et des N-TEF permet la transition vers une élongation productive (voir texte pour plus de détails). |
Plus récemment, des études réalisées à l’échelle du génome ont permis de mesurer l’étendue de ce phénomène. Ainsi, la pause transcriptionnelle de l’ARN pol II est détectée à proximité de nombreux promoteurs de gènes humains [2]. L’expression des gènes n’est donc pas seulement régulée au cours de l’initiation via le recrutement de l’ARN pol II au niveau du promoteur, elle est également contrôlée par la capacité de l’ARN pol II à entrer en phase d’élongation dite « productive ». L’utilisation d’inhibiteurs spécifiques de CDK9 a mis en évidence le rôle central de P-TEFb dans ce mécanisme. Dans le contexte cellulaire, l’activité de CDK9 doit donc être finement contrôlée. En 2001, deux études indépendantes ont mis en évidence un système particulier de régulation de l’activité de P-TEFb [4, 5]. Un petit ARN non codant, l’ARN 7SK, s’associe à P-TEFb pour moduler son activité kinase. Cet ARN est l’un des premiers exemples d’ARN non codant capable de moduler la transcription par l’ARN pol II [6].
Contrôle de l’activité de P-TEFb par la RNP 7SK/HEXIM
L’ARN 7SK est un petit ARN nucléaire de 331 nucléotides, conservé chez les métazoaires [6]. Deux protéines, MePCE (methylphosphate capping enzyme) et LARP7 (La related protein, member 7), s’associent à l’ARN 7SK pour protéger ses extrémités, formant ainsi la ribonucléoparticule (RNP) 7SK « cœur ». MePCE (également appelée BCDIN3) ajoute la coiffe à l’extrémité 5’ de l’ARN, alors que LARP7 (également appelée PIP7S) est une protéine de type La qui lie la région 3’ de 7SK [6–9]. Cette RNP « cœur » joue un rôle de plateforme structurale permettant la liaison d’autres protéines. La protéine HEXIM1 (et/ou HEXIM2- hexamethylene bis-acetamide inducible 1 ou 2) s’associe sous forme de dimère à une structure en tige/boucle située à l’extrémité 5’ de l’ARN 7SK [10]. La liaison à l’ARN 7SK entraîne un changement conformationnel des protéines HEXIM leur permettant d’interagir avec la cycline T1 [11]. P-TEFb se retrouve alors séquestré dans un complexe ribonucléoprotéique au sein duquel l’activité kinase de CDK9 est inhibée. Dans les cellules HeLa, environ 50 % de P-TEFb est maintenu sous forme inactive, associé à la RNP 7SK/HEXIM [4]. Deux populations de P-TEFb coexistent donc dans la cellule : la forme libre et active capable de stimuler la transcription, et la forme inactive séquestrée par l’ARN 7SK (Figure 1). La forme libre de P-TEFb est recrutée au niveau des gènes transcrits par divers facteurs de transcription tels que Brd4, NF-kB, c-Myc ou MyoD [1].
L’association d’HEXIM et de P-TEFb à la RNP 7SK « cœur » est dynamique et réversible, et peut être modulée par les conditions environnementales [4, 5]. Par exemple, l’inhibition de la transcription induit la dissociation du complexe P-TEFb/7SK/HEXIM, permettant une rapide augmentation de la quantité de molécules P-TEFb actives et disponibles pour la transcription par l’ARN pol II. Pour empêcher sa réassociation avec HEXIM et P-TEFb, l’ARN 7SK libéré est, quant à lui, titré par des protéines de type hnRNP (heterogeneous nuclear ribonucleoproteins) [12, 13]. Ce mécanisme de régulation consiste donc à moduler la quantité de P-TEFb disponible pour activer la transcription. La forme inactive peut ainsi être considérée comme un réservoir cellulaire de P-TEFb, mobilisable à tout moment par la cellule.
L’équilibre dynamique entre les formes active et inactive de P-TEFb permet d’adapter finement le taux de P-TEFb disponible en fonction des besoins transcriptionnels de la cellule. Tout dysfonctionnement ou stimulus capable de modifier cet équilibre peut profondément perturber l’activité physiologique de la cellule. La dérégulation de l’activité de P-TEFb peut, en effet, conduire à d’importantes modifications de l’expression génique, et être à l’origine de pathologies graves. La RNP 7SK « cœur » et les protéines HEXIM ont d’ailleurs un rôle désormais bien établi dans des pathologies telles que l’hypertrophie cardiaque, le syndrome d’immunodéficience acquise (SIDA) ou certains cancers, ce qui souligne l’importance de ce système de régulation de l’activité de P-TEFb.
Dérégulation de l’activité de P-TEFb et hypertrophie cardiaque
L’hypertrophie cardiaque se caractérise par un élargissement global de la taille des cardiomyocytes à la suite d’une augmentation des taux d’ARN et de protéines cellulaires. Cette réponse représente normalement une adaptation physiologique à un travail accru du cœur, mais expose, lorsqu’elle persiste, à un risque élevé de défaillance cardiaque. Les études qui portent sur les mécanismes moléculaires à l’origine de la maladie chez l’homme soulignent l’importance de la régulation de l’activité de P-TEFb par la RNP 7SK/HEXIM dans les cardiomyocytes [14]. Divers signaux hypertrophiques, certains facteurs de croissance ou un stress mécanique sévère, activent des voies de signalisation qui influencent directement l’équilibre entre les formes active et inactive de P-TEFb. Ces signaux hypertrophiques induisent la dissociation du complexe inactif, augmentant le taux de P-TEFb actif dans des cellules où l’activité de CDK9 est normalement limitante. L’activation de P-TEFb stimule alors la transcription par l’ARN pol II, entraînant l’augmentation du taux d’ARNm et de protéines cellulaires. En accord avec une stimulation de l’élongation de la transcription, le niveau de phosphorylation du CTD, et notamment celui de la Ser2, augmente dans les conditions de stress hypertrophique.
Le contrôle de la disponibilité de P-TEFb est donc un mécanisme essentiel pour la fonction et le développement des cardiomyocytes. Chez la souris, l’activation aberrante de P-TEFb engendre, comme chez l’homme, des manifestations d’hypertrophie cardiaque. Par exemple, la délétion génétique de CLP-1, l’homologue murin d’HEXIM1, entraîne une létalité embryonnaire qu’explique un élargissement pathologique du cœur mimant les caractéristiques de l’hypertrophie cardiaque [15]. De la même façon, l’inactivation de l’ARN 7SK par ARN interférence dans les cardiomyocytes induit une activation de CDK9, suffisante pour induire une croissance anormale de ces cellules. Au-delà de l’élargissement des cellules, l’activation constitutive de CDK9 aboutit également à un dysfonctionnement des mitochondries [14] qui entraîne l’apoptose des myocytes et prédispose à la défaillance cardiaque.
Dans les cardiomyocytes, la régulation de P-TEFb par la RNP 7SK/HEXIM permet de limiter l’activité de CDK9. Les signaux hypertrophiques perturbent l’équilibre entre forme active et inactive de P-TEFb et font de l’hypertrophie cardiaque un modèle physiopathologique idéal pour étudier le système de régulation de P-TEFb. L’étude approfondie des cascades de signalisation activées par les signaux hypertrophiques, et notamment celles qui sont dépendantes du calcium (calcineurine) et Jak/STAT [14], pourrait permettre de mieux comprendre les mécanismes moléculaires qui régissent la séquestration de P-TEFb par la RNP 7SK/HEXIM.
Le système de régulation de P-TEFb, cible du VIH-1
Comme tous les rétrovirus, le virus de l’immunodéficience humaine de type 1 (VIH-1) intègre son génome dans celui de la cellule infectée afin de se répliquer. L’expression des gènes viraux est alors contrôlée par la machinerie transcriptionnelle de la cellule hôte. La transcription basale à partir du promoteur LTR (long terminal repeat) du VIH est inefficace, car la progression de l’ARN pol II sur le génome viral est bloquée par les N-TEF peu après l’initiation. La transcription est activée par une protéine virale appelée Tat, qui facilite le passage vers une élongation productive [16]. Pour cela, Tat recrute P-TEFb au niveau de l’ARN pol II bloquée en formant un complexe stable avec la cycline T1 et une structure en tige/boucle appelée TAR (trans-acting response element), formée à l’extrémité 5’ de l’ARNm viral naissant [17]. P-TEFb peut ensuite stimuler la reprise de la transcription et la production de transcrits pleine-taille. Il a récemment été montré que le complexe P-TEFb recruté au niveau de l’ARN TAR est associé aux autres protéines du super elongation complex (SEC) [3, 18–20]. L’association de Tat avec le SEC permet donc le recrutement simultané de plusieurs facteurs d’élongation au niveau du promoteur viral.
L’activité kinase de CDK9 est essentielle et limitante pour la transcription des gènes viraux, qui semble plus sensible à la disponibilité de P-TEFb que les promoteurs cellulaires [21]. Ainsi, tous les inhibiteurs de P-TEFb identifiés à ce jour inhibent efficacement la réplication du virus. Un taux restreint de P-TEFb actif, associé à la mise en place d’une structure chromatinienne restrictive au niveau du promoteur LTR, semble être à l’origine de l’établissement de la latence du virus dans les cellules T CD4+ primaires [22].
Dans les cellules dans lesquelles le VIH se réplique, l’équilibre entre les formes active et inactive de P-TEFb est modifié. En effet, la protéine Tat, même en l’absence d’autres protéines et ARN viraux, est capable de libérer la fraction de P-TEFb séquestrée dans la RNP 7SK/HEXIM, entraînant ainsi une augmentation significative du taux de P-TEFb libre dans la cellule [23, 24]. Comment le VIH, via la protéine Tat, détourne-t-il le système de régulation de P-TEFb endogène ? Deux cibles principales de Tat ont été identifiées : l’ARN 7SK et la cycline T1. Tat se lie à l’ARN 7SK au niveau du site de liaison d’HEXIM [25], créant une compétition entre ces deux protéines pour la liaison à l’ARN 7SK [24, 25]. La détection d’un complexe contenant Tat, l’ARN 7SK, P-TEFb mais pas HEXIM, suggère que Tat peut, au moins transitoirement, remplacer HEXIM au sein de la RNP 7SK/P-TEFb (Figure 2) [20]. De plus, Tat interagit directement avec la cycline T1 et entre en compétition avec HEXIM pour la liaison à P-TEFb [20, 23]. La plus forte affinité de Tat pour la cycline T1 bloque l’interaction entre HEXIM1 et P-TEFb [15, 23], empêchant ainsi la séquestration de P-TEFb au sein de la RNP 7SK/HEXIM. Une autre étude suggère que Tat s’associe également avec la forme inactive de P-TEFb. Ce complexe est alors recruté au promoteur du VIH avant l’initiation de la transcription, via son association avec le complexe de pré-initiation, de manière indépendante de l’ARN TAR (Figure 2). Après l’initiation, l’ARN TAR naissant permettrait la dissociation de la RNP 7SK/HEXIM [26]. Le site de liaison de Tat à l’ARN TAR étant structurellement identique à celui présent dans l’ARN 7SK [25], une compétition entre les deux ARN pour la liaison de Tat pourrait donc induire l’activation de P-TEFb. Malgré tout, le niveau de la RNP 7SK/HEXIM détecté sur le promoteur LTR est très faible et ne peut pas expliquer le taux élevé de P-TEFb observé lors de la transactivation par Tat. Il est par conséquent probable que les deux mécanismes qui recrutent P-TEFb fonctionnent en synergie pour activer la transcription des gènes viraux.
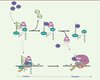 |
Figure 2. Libération de la fraction inactive de P-TEFb par la protéine virale Tat. Tat lie l’ARN 7SK au niveau du site de liaison d’HEXIM1 et remplace HEXIM1 au sein d’un complexe contenant P-TEFb et la RNP 7SK « coeur ». Après relargage de P-TEFb, une autre protéine Tat s’associe à la cycline T1 et recrute P-TEFb au niveau des gèlnes viraux, via la liaison à l’ARN TAR. Au sein du SEC, P-TEFb active la transcription du génome viral. La forme inactive de P-TEFb peut également être recrutée par Tat directement au niveau du promoteur LTR. |
La protéine Tat joue donc un double rôle dans l’activation transcriptionnelle des gènes viraux. D’une part, elle recrute le SEC au niveau de la machinerie transcriptionnelle via l’ARN TAR. D’autre part, elle modifie le taux intracellulaire de P-TEFb actif en perturbant son système de régulation endogène, de façon à obtenir la concentration en P-TEFb nécessaire à la transcription de ses propres gènes.
P-TEFb et cancer
L’équilibre entre les formes active et inactive de P-TEFb est adapté à l’état de croissance et de différenciation de la cellule [1]. Une modification de cet équilibre peut engendrer d’importantes perturbations de l’expression génique, pouvant entraîner une prolifération non contrôlée des cellules. Ainsi, un niveau élevé de P-TEFb a été mis en évidence dans différents types de tumeurs [27]. Par ailleurs, une surexpression de la cycline T1 peut induire la transformation de cellules in vitro et une accélération du développement de tumeurs chez la souris, soulignant le rôle oncogénique de P-TEFb [27].
L’activation de P-TEFb peut également résulter d’une dérégulation ou d’une mutation affectant l’un des constituants de la RNP 7SK/HEXIM ou son intégrité. Une diminution de l’expression d’HEXIM1 a ainsi été observée dans les cancers du sein par rapport aux niveaux observés dans des tissus mammaires sains [15]. En effet, dans les cellules mammaires, la surexpression d’HEXIM1 bloque le recrutement de P-TEFb et du récepteur aux œstrogènes a (REa) sur le promoteur de ses gènes cibles, impliqués dans la prolifération cellulaire et l’apoptose. HEXIM1 inhibe donc l’expression de ces gènes, limitant ainsi la prolifération des cellules mammaires [28]. La présence d’HEXIM1 au niveau des promoteurs des gènes cibles du REa est d’ailleurs un élément clé de la réponse au tamoxifène, un antagoniste du récepteur des œstrogènes utilisé dans le traitement des cancers du sein œstrogéno-dépendants [29].
La protéine LARP7, quant à elle, assure l’intégrité des RNP 7SK en stabilisant l’ARN 7SK. La déplétion de LARP7 entraîne celle de l’ARN 7SK, avec pour conséquence une augmentation du niveau de P-TEFb actif dans les cellules [7]. Chez l’homme, des mutations dans la partie carboxy-terminale de LARP7, identifiées dans des tumeurs gastriques, abolissent son interaction avec l’ARN 7SK et affectent l’intégrité de la RNP 7SK/HEXIM/P-TEFb [7]. De la même façon, la déplétion de LARP7 dans des cellules épithéliales mammaires en culture entraîne leur transformation, un processus qui requiert la hausse d’activité de P-TEFb [7]. La diminution de l’expression de LARP7 semble également être un bon marqueur prédictif de la présence de métastases ganglionnaires à un stade tumoral précoce [30]. L’activation de P-TEFb, qui résulte d’une déstabilisation de la RNP 7SK/HEXIM/P-TEFb, pourrait donc jouer un rôle important dans la prolifération et l’agressivité des cellules cancéreuses.
Par ailleurs, trois sous-unités du SEC ont été identifiées comme partenaires de translocation de la protéine MLL (mixed lineage leukemia) [31]. Ces protéines, lorsqu’elles sont fusionnées au domaine de liaison à l’ADN de MLL, recrutent le SEC au niveau des gènes cibles de MLL et modifient leur expression. Il peut en résulter une prolifération non contrôlée des progéniteurs hématopoïétiques et une transformation leucémique. La présence de P-TEFb au sein du SEC avec les partenaires de translocation de la protéine MLL est un argument en faveur de son implication dans le développement des leucémies infantiles. Ainsi, l’efficacité thérapeutique du flavopiridol, un composé antiprolifératif qui cible spécifiquement l’activité de CDK9 dans certaines leucémies, est actuellement testée dans des essais cliniques. Le contrôle du système de régulation endogène de P-TEFb pourrait donc représenter une solution alternative pour l’élaboration de nouvelles drogues anticancéreuses.
Conclusion
La découverte de la fonction cellulaire de l’ARN 7SK, en 2001, a constitué une avancée significative dans la compréhension des mécanismes moléculaires contrôlant l’activité de P-TEFb dans les cellules humaines. Les recherches entreprises depuis 10 ans ont permis de mieux caractériser la RNP 7SK/HEXIM et son mode d’action. Ainsi, certains stress, certaines protéines ou molécules qui modifient le taux de P-TEFb actif en perturbant son système de régulation par la RNP 7SK/HEXIM ont été identifiés (Figure 3). De nombreuses questions restent toutefois en suspens. Quels mécanismes régissent l’équilibre dynamique entre les formes active et inactive de P-TEFb ? Quelles cascades de signalisation libèrent P-TEFb de la RNP 7SK/HEXIM dans le cas de l’hypertrophie cardiaque ? Serait-il possible de bloquer ou limiter la réplication du VIH en empêchant la dissociation de la RNP 7SK/HEXIM/P-TEFb ? Quel rôle joue la RNP 7SK/HEXIM dans l’établissement de la latence du VIH ? Les études qui portent sur ces pathologies ont déjà contribué à la découverte de P-TEFb et de son système de régulation. Elles pourraient désormais ouvrir de nouvelles perspectives dans le développement d’approches thérapeutiques visant à cibler spécifiquement l’activité kinase de P-TEFb.
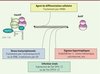 |
Figure 3. Équilibre dynamique entre les formes active et inactive de P-TEFb. Un stress transcriptionnel, l’infection par le VIH ou des signaux hypertrophiques peuvent induire la dissociation de la RNP 7SK/HEXIM et libérer la forme active de P-TEFb. L’équilibre peut également être déplacé vers la forme inactive lorsque l’on induit la différenciation cellulaire. |
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Références
- Zhou Q, Yik JH. The Yin and Yang of P-TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol Mol Biol Rev 2006 ; 70 : 646–659. [CrossRef] [PubMed] [Google Scholar]
- Nechaev S, Adelman K. Pol II waiting in the starting gates: regulating the transition from transcription initiation into productive elongation. Biochim Biophys Acta 2011 ; 1809 : 34–45. [PubMed] [Google Scholar]
- He N, Zhou Q. New insights into the control of HIV-1 transcription: when Tat meets the 7SK snRNP and super elongation complex (SEC). J Neuroimmune Pharmacol 2011 ; 6 : 260–268. [CrossRef] [PubMed] [Google Scholar]
- Nguyen VT, Kiss T, Michels AA, Bensaude O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 2001 ; 414 : 322–325. [CrossRef] [PubMed] [Google Scholar]
- Yang Z, Zhu Q, Luo K, Zhou Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 2001 ; 414 : 317–322. [CrossRef] [PubMed] [Google Scholar]
- Diribarne G, Bensaude O. 7SK RNA, a non-coding RNA regulating P-TEFb, a general transcription factor. RNA Biol 2009 ; 6 : 122–128. [CrossRef] [PubMed] [Google Scholar]
- He N, Jahchan NS, Hong E, et al. A La-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol Cell 2008 ; 29 : 588–599. [CrossRef] [PubMed] [Google Scholar]
- Jeronimo C, Forget D, Bouchard A, et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell 2007 ; 27 : 262–274. [CrossRef] [PubMed] [Google Scholar]
- Xue Y, Yang Z, Chen R, Zhou Q. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res 2010 ; 38 : 360–369. [CrossRef] [PubMed] [Google Scholar]
- Egloff S, Van Herreweghe E, Kiss T. Regulation of polymerase II transcription by 7SK snRNA: two distinct RNA elements direct P-TEFb and HEXIM1 binding. Mol Cell Biol 2006 ; 26 : 630–642. [CrossRef] [PubMed] [Google Scholar]
- Michels AA, Fraldi A, Li Q, et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. Embo J 2004 ; 23 : 2608–2619. [CrossRef] [PubMed] [Google Scholar]
- Barrandon C, Bonnet F, Nguyen VT, et al. The transcription-dependent dissociation of P-TEFb-HEXIM1–7SK RNA relies upon formation of hnRNP-7SK RNA complexes. Mol Cell Biol 2007 ; 27 : 6996–7006. [CrossRef] [PubMed] [Google Scholar]
- Van Herreweghe E, Egloff S, Goiffon I, et al. Dynamic remodelling of human 7SK snRNP controls the nuclear level of active P-TEFb. EMBO J 2007 ; 26 : 3570–3580. [CrossRef] [PubMed] [Google Scholar]
- Sano M, Schneider MD. Cyclin-dependent kinase-9: an RNAPII kinase at the nexus of cardiac growth and death cascades. Circ Res 2004 ; 95 : 867–876. [CrossRef] [PubMed] [Google Scholar]
- Dey A, Chao SH, Lane DP. HEXIM1 and the control of transcription elongation: from cancer and inflammation to AIDS and cardiac hypertrophy. Cell Cycle 2007 ; 6 : 1856–1863. [CrossRef] [PubMed] [Google Scholar]
- Cherrier T, Le Douce V, Redel L, et al. Un virus tapi dans l’ombre : les bases moléculaires de la latence du VIH-1. Partie II : la réactivation de la latence du VIH-1 et ses implications thérapeutiques. Med Sci (Paris) 2010 ; 26 : 291–295. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Wang Y, Liu XY, De Clercq E. Role of the HIV-1 positive elongation factor P-TEFb and inhibitors thereof. Mini Rev Med Chem 2009 ; 9 : 379–385. [PubMed] [Google Scholar]
- He N, Liu M, Hsu J, et al. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell 2010 ; 38 : 428–438. [CrossRef] [PubMed] [Google Scholar]
- Lin C, Smith ER, Takahashi H, et al. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 2010 ; 37 : 429–437. [CrossRef] [PubMed] [Google Scholar]
- Sobhian B, Laguette N, Yatim A, et al. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell 2010 ; 38 : 439–451. [CrossRef] [PubMed] [Google Scholar]
- Flores O, Lee G, Kessler J, et al. Host-cell positive transcription elongation factor b kinase activity is essential and limiting for HIV type 1 replication. Proc Natl Acad Sci USA 1999 ; 96 : 7208–7213. [CrossRef] [Google Scholar]
- Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol 2010 ; 84 : 6425–6437. [CrossRef] [PubMed] [Google Scholar]
- Barboric M, Yik JH, Czudnochowski N, et al. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res 2007 ; 35 : 2003–2012. [CrossRef] [PubMed] [Google Scholar]
- Sedore SC, Byers SA, Biglione S, et al. Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res 2007 ; 35 : 4347–4358. [CrossRef] [PubMed] [Google Scholar]
- Muniz L, Egloff S, Ughy B, et al. Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog 2010 ; 6 : e1001152. [CrossRef] [PubMed] [Google Scholar]
- D’Orso I, Frankel AD. RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat Struct Mol Biol 2010 ; 17 : 815–821. [Google Scholar]
- Moiola C, De Luca P, Gardner K, et al. Cyclin T1 overexpression induces malignant transformation and tumor growth. Cell Cycle 2010 ; 9 : 3119–3126. [CrossRef] [PubMed] [Google Scholar]
- Ogba N, Chaplin LJ, Doughman YQ, et al. HEXIM1 regulates 17beta-estradiol/estrogen receptor-alpha-mediated expression of cyclin D1 in mammary cells via modulation of P-TEFb. Cancer Res 2008 ; 68 : 7015–7024. [CrossRef] [PubMed] [Google Scholar]
- Ketchart W, Ogba N, Kresak A, et al. HEXIM1 is a critical determinant of the response to tamoxifen. Oncogene 2011 ; 30 : 3563–3569. [CrossRef] [PubMed] [Google Scholar]
- Biewenga P, Buist MR, Moerland PD, et al. Gene expression in early stage cervical cancer. Gynecol Oncol 2008 ; 108 : 520–526. [CrossRef] [PubMed] [Google Scholar]
- Smith E, Lin C, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev 2011 ; 25 : 661–672. [Google Scholar]
Liste des figures
|
Figure 1. Contrôle de l’activité de P-TEFb et élongation de la transcription. P-TEFb existe sous deux formes en équilibre dynamique dans les cellules humaines. Une fraction de P-TEFb est inactivée par l’ARN 7SK et l’autre fraction est sous forme libre et active. La forme active est recrutée au niveau de l’ARN pol II au sein du super elongation complex (SEC) pour lever la pause transcriptionnelle imposée par les facteurs négatifs d’élongation DSIF et NELF. La phosphorylation par P-TEFb du CTD de l’ARN pol II et des N-TEF permet la transition vers une élongation productive (voir texte pour plus de détails). |
| Dans le texte | |
|
Figure 2. Libération de la fraction inactive de P-TEFb par la protéine virale Tat. Tat lie l’ARN 7SK au niveau du site de liaison d’HEXIM1 et remplace HEXIM1 au sein d’un complexe contenant P-TEFb et la RNP 7SK « coeur ». Après relargage de P-TEFb, une autre protéine Tat s’associe à la cycline T1 et recrute P-TEFb au niveau des gèlnes viraux, via la liaison à l’ARN TAR. Au sein du SEC, P-TEFb active la transcription du génome viral. La forme inactive de P-TEFb peut également être recrutée par Tat directement au niveau du promoteur LTR. |
| Dans le texte | |
|
Figure 3. Équilibre dynamique entre les formes active et inactive de P-TEFb. Un stress transcriptionnel, l’infection par le VIH ou des signaux hypertrophiques peuvent induire la dissociation de la RNP 7SK/HEXIM et libérer la forme active de P-TEFb. L’équilibre peut également être déplacé vers la forme inactive lorsque l’on induit la différenciation cellulaire. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.