")
")
| Issue |
Med Sci (Paris)
Volume 28, Number 2, Février 2012
|
|
|---|---|---|
| Page(s) | 130 - 133 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2012282004 | |
| Published online | 27 février 2012 | |
Développement embryonnaire précoce
Un grand rôle pour un petit ARN
Extreme microcephaly and growth retardation caused by mutations in a non-coding RNA component of the minor spliceosome
1
Hospices civils de Lyon, groupement hospitalier Est, hôpital femme-mère-enfant, 6 e étage, 59, boulevard Pinel, 69677 Bron Cedex, France
2
UMR CNRS 5292
3
Inserm U1068
4
Inserm U781
5
Université Paris Descartes, France
6
Hôpital Necker-Enfants malades, 149, rue de Sèvres 75743 Paris Cedex 15, France
7
Inserm, U946
8
Université Paris Diderot, Sorbonne Paris Cité, 27 rue Juliette Dodu, 75010 Paris, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Le splicéosome est un complexe ribonucléoprotéique qui catalyse l’épissage des ARN prémessagers situés dans le noyau en réalisant l’excision des introns puis la ligation des exons pour produire des ARN messagers (ARNm) matures, qui sont ensuite exportés dans le cytoplasme pour être traduits en protéines. Ce complexe est composé de cinq ribonucléoprotéines (small nuclear RNP, snRNP) constituées de petits ARN nucléaires (snARN) et de protéines. Notre équipe a récemment montré pour la première fois le rôle d’un petit ARN du splicéosome, U4atac snARN, dans le développement embryonnaire et la survie postnatale chez l’homme [1].
Stratégie d’identification du petit ARN nucléaire U4atac responsable du syndrome de Taybi-Linder
Le syndrome de Taybi-Linder (TALS, MIM 210710) ou nanisme microcéphalique primordial de type 1 (MOPD1) est un syndrome malformatif rare, autosomique récessif, dont l’étiologie était jusqu’à présent inconnue. Ce syndrome a été décrit en 1967 [2] et à ce jour, environ 50 cas ont été rapportés dans la littérature médicale et scientifique (P. Edery et al., en préparation). Le MOPD1/TALS est caractérisé par un retard de croissance intra-utérin et un retard statural postnatal importants, des os longs incurvés et brefs avec un retard de maturation très marqué des épiphyses, des articulations fixées en flexion, une microcéphalie très sévère et des malformations cérébrales à type d’agyrie ou pachygyrie1, une dysmorphie faciale caractéristique, une peau sèche et des cheveux rares et fins (Figure 1A-C). Une dégradation rapide et inexpliquée de l’état général et neurologique, qui évolue vers le décès en quelques jours, survient en général avant l’âge de trois ans. En 2006, notre équipe a localisé le gène responsable du MOPD1/TALS dans une région de 13 cM sur le chromosome 2q14 grâce au génotypage de quatre malades appartenant à trois familles consanguines originaires du bassin méditerranéen, avec des marqueurs polymorphes couvrant l’ensemble du génome. Dans cette étude [3], la localisation génétique a été effectuée grâce au développement d’une nouvelle méthode de cartographie par homozygotie utilisant le coefficient de consanguinité réel des malades, estimé d’après leur information génotypique plutôt que d’après les informations généalogiques, qui étaient insuffisantes et imprécises. Nous avons ensuite restreint l’intervalle candidat à une région de 3,19 Mb grâce à une quatrième famille consanguine d’origine méditerranéenne. Le séquençage classique par la méthode de Sanger n’a révélé aucune mutation dans les séquences codantes et introniques flanquantes des gènes situés dans le nouvel intervalle candidat. Nous avons alors effectué, en collaboration avec la société IntegraGen, un séquençage haut débit ciblé de la totalité de la région candidate (capture Agilent SureSelectTM et séquençage Illumina GAII) chez 10 individus des quatre familles étudiées. Le choix attentif préalable des individus chez lesquels nous séquencerions la région candidate nous a permis d’appliquer les filtres qui ont conduit à l’identification du gène responsable du MOPD1/TALS. Dans deux familles, nous avons sélectionné un enfant atteint (autozygote pour l’haplotype associé à la maladie) et un individu sain de la fratrie ne portant pas cet haplotype, de façon à pouvoir reconstruire les haplotypes des deux parents. Dans les deux autres familles, dans lesquelles il n’existait pas de germain sain non porteur d’un haplotype associé à la maladie, nous avons sélectionné un enfant atteint et ses deux parents (hétérozygotes). Parmi les variations nucléotidiques identifiées, 4 356 différaient de la séquence génomique de référence, dont 1 015 n’étaient pas référencées dans les bases de données dbSNP et du NCBI (National center for biotechnology Information). Parmi celles-ci, une seule substitution G > A était retrouvée à la fois à l’état homozygote chez les quatre individus atteints non apparentés et à l’état hétérozygote chez les parents. La mutation récurrente G > A est située en position 51 de la séquence non codante RNU4ATAC, transcrite en un petit ARN fonctionnel du splicéosome appelé U4atac [4, 5]. Le séquençage classique par la méthode de Sanger effectué chez tous les individus des quatre familles nucléaires étudiées a confirmé la concordance de la cotransmission de la mutation g.51G>A du snARN U4atac avec la maladie. Trois autres mutations du snARN U4atac ont ensuite été identifiées chez les patients de quatre autres familles MOPD1/TALS, confirmant le rôle de ce petit ARN dans la pathologie.
 |
Figure 1. Phénotype du MOPD1/TALS. A, B. Patient en période néonatale. A. On note un visage rond, un occiput proéminent, une protrusion oculaire, un nez busqué, un menton petit et en retrait, un cou court, une peau sèche, une absence de cheveux, des contractures articulaires des genoux et des coudes ainsi que des mains et pieds courts. B. Radiographies osseuses montrant, aux membres inférieurs, un retard d’ossification des épiphyses tibiales supérieures et fémorales inférieures (les flèches vertes indiquent la position attendue des épiphyses ossifiées, absentes chez le patient), des fémurs courts et incurvés et des métaphyses fémorales discrètement élargies (flèche rouge) et, au niveau de la colonne, un discret aplatissement des corps vertébraux (platyspondylie) et une cyphose lombaire (indiquée par le crochet blanc). C. Imagerie cérébrale par résonance magnétique (IRM) à l’âge de 1 mois. Kyste arachnoïdien (flèche blanche), hypoplasie cérébrale et pachygyrie (flèche jaune), agénésie de la partie antérieure du corps calleux (la pointe de la tête de flèche rouge montre la position attendue de la partie antérieure du corps calleux, ici absente) et discrète hypoplasie du vermis cérébelleux (flèche bleue). |
snARN U4atac et son rôle dans le splicéosome mineur et le processus d’épissage
Le snARN U4atac forme une bimolécule avec le snARN U6atac (appariement par des nucléotides complémentaires) au cours du processus d’épissage. Au sein de ce duplex, la tige I et la tige II du snARN U4atac, qui présentent un appariement nucléotidique intermoléculaire avec le snARN U6atac, sont séparées par une région tige-boucle formée par des appariements nucléotidiques intramoléculaires (Figure 2). Les quatre mutations identifiées dans notre étude sont situées dans cette région tige-boucle du snARN U4atac. La modélisation in silico de cette tige-boucle montre que les mutations identifiées modifient la conformation bidimensionnelle du site de fixation d’une protéine appelée 15.5K, elle-même nécessaire à la fixation de prp31, une protéine importante pour la stabilisation du tri-snRNP U4atac/U6atac.U5 et le processus d’épissage. Les mutations identifiées, g.51G>A, g.50G>A, g.50G>C et g.53C>G du snARN U4atac sont situées à des positions nucléotidiques relativement bien conservées entre les espèces. Toutefois, chez certaines espèces comme le poisson zèbre, la séquence g.51G>A est constitutive. Dans ces cas, il existe une « mutation compensatoire » du nucléotide apparié, qui restaure la structure bidimensionnelle de la tige-boucle du snARN U4atac. Ce type d’observation indique que la conservation entre les espèces de la structure bidimensionnelle du snARN U4atac est plus forte que celle des nucléotides qui le composent. Le snRNP U4atac fait partie d’un complexe ribonucléoprotéique appelé splicéosome mineur ou U12, composé également des snRNP U6atac, U5, U11 et U12.
 |
Figure 2. Modèle bidimensionnel de la bimolécule humaine formée des snARN U4atac/U6atac. La tige boucle située en 5’ du snRNA U4atac résulte d’un appariement intramoléculaire entre nucléotides complémentaires. Les mutations identifiées chez les patients MOPD1/TALS sont situées le plus souvent dans la tige boucle 5’ du snARN U4atac. Le site de fixation de la protéine 15.5K, indiqué par un disque bleu, est nécessaire à l’association avec le snARN U5 (adapté de [10] et [11]). |
Le splicéosome U12 permet l’épissage d’environ 700 à 800 introns dans le génome, les autres introns du génome, majoritaires, étant épissés par le splicéosome majeur [6]. Les introns reconnus par le splicéosome U12, ou introns U12, sont caractérisés par leurs séquences consensus d’épissage, en particulier leur site 5’ d’épissage et leur site de branchement [7]. La plupart des gènes qui contiennent un ou plusieurs introns U12, ou gènes U12, sont impliqués dans des fonctions cellulaires clés comme la réplication de l’ADN, la transcription, la maturation des ARN et leur transport, la traduction et l’organisation du cytosquelette de la cellule. Certains gènes U12 appartiennent à des familles de canaux ioniques et un petit nombre d’entre eux ont un rôle probable dans le développement embryonnaire osseux ou cérébral. Par des études quantitatives des niveaux d’expression de transcrits, nous avons montré que la présence à l’état homozygote de la mutation récurrente g.51G>A du snARN U4atac entraîne un défaut de l’épissage de tous les introns U12 étudiés, sans l’abolir complètement (quantité accrue d’introns U12 non épissés dans les transcrits fibroblastiques des patients par rapport aux individus contrôles). Les mutations du snARN U4atac identifiées à ce jour chez les patients MOPD1/TALS entraînent donc une rétention des introns U12 dans les ARNm sans abolir complètement l’épissage U12 (effet hypomorphe, Figure 3). De plus, le niveau d’expression de certains transcrits U12 était abaissé, posant la question du mécanisme de dégradation de ces transcrits aberrants. Toutefois, le niveau d’expression de la plupart des transcrits U12 étudiés était normal ou même augmenté. Ces résultats étaient très reproductibles mais ils restent inexpliqués à ce jour. En particulier, le transcrit du gène PHB2, impliqué dans des fonctions mitochondriales, était très abaissé dans les fibroblastes des patients de notre étude. Il est intéressant de noter qu’une étude récente [8] a montré que le transcrit Phb2 (prohibitin 2) était également abaissé chez la larve de drosophile mutante, knock-out pour le snARN U6atac, partenaire de U4atac, ce qui suggère un rôle possible du gène PHB2 - ou des gènes impliqués dans cette voie de signalisation - dans le phénotype MOPD1/TALS. Indépendamment, une autre équipe a identifié la mutation g.51G>A du snARN U4atac chez des patients MOPD1/TALS Amish (isolat d’une population de l’Ohio, États-Unis) et trois autres mutations chez deux autres patients, confirmant le rôle du snARN U4atac dans ce syndrome [9].
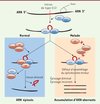 |
Figure 3. Splicéosome mineur : une fonction vitale. Les défauts de l’épissage des introns de type U12 sont dus à un défaut de la fonction du splicéosome mineur (U12-dépendant). Les mutations MOPD1/TALS conduisent à une accumulation des transcrits prémessagers non épissés ; 700 à 800 gènes humains contenant des introns U12 ont probablement un niveau d’expression plus ou moins modifié (démontré dans notre étude [1] pour certains d’entre eux) (d’après [12]). |
Ces découvertes permettent de formuler des hypothèses sur le rôle possible de nouveaux gènes (U12 ou leurs cibles) dans le développement osseux et cérébral et la survie postnatale. Elles rendent également possible, dans un avenir proche, l’observation directe du rôle du splicéosome mineur dans l’épissage de l’ensemble des introns U12 du génome, grâce au séquençage massif de nouvelle génération du transcriptome des cellules des patients MOPD1/TALS.
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Nous remercions chaleureusement les familles qui ont contribué à cette étude. Ce travail a été financé par la Fondation Yves Cotrel-Institut de France, les Hospices civils de Lyon, le ministère de la Santé et des Solidarités (Direction de l’hospitalisation et de l’organisation des soins, plan maladies rares 2004-2008), l’université Lyon 1, l’Inserm et l’association GEN’HOM.
Absence (agyrie) ou diminution (pachygyrie) des circonvolutions cérébrales aboutissant à une surface cérébrale lisse.
Références
- Edery P, Marcaillou C, Sahbatou M, et al. Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science 2011 ; 332 : 240–243. [CrossRef] [PubMed] [Google Scholar]
- Taybi H, Linder D. Congenital familial dwarfism with cephaloskeletal dysplasia. Radiology 1967 ; 89 : 275–281. [CrossRef] [Google Scholar]
- Leutenegger AL, Labalme A, Genin E, et al. Using genomic inbreeding coefficient estimates for homozygosity mapping of rare recessive traits: application to Taybi-Linder syndrome. Am J Hum Genet 2006 ; 79 : 62–66. [CrossRef] [PubMed] [Google Scholar]
- Tarn WY, Steitz JA. Highly diverged U4 and U6 small nuclear RNAs required for splicing rare AT-AC introns. Science 1996 ; 273 : 1824–1832. [CrossRef] [PubMed] [Google Scholar]
- Tarn WY, Steitz JA. A novel spliceosome containing U11, U12, and U5 snRNPs excises a minor class (AT-AC) intron in vitro. Cell 1996 ; 84 : 801–811. [CrossRef] [PubMed] [Google Scholar]
- Alioto TS. U12DB: a database of orthologous U12-type spliceosomal introns. Nucleic Acids Res 2007; 35 : D110–D115. [CrossRef] [PubMed] [Google Scholar]
- Sharp PA, Burge CB. Classification of introns: U2-type or U12-type. Cell 1997 ; 91 : 875–879. [CrossRef] [PubMed] [Google Scholar]
- Pessa HK, Greco D, Kvist J, et al. Gene expression profiling of U12-type spliceosome mutant Drosophila reveals widespread changes in metabolic pathways. PLoS One 2010 ; 5 : e13215. [CrossRef] [PubMed] [Google Scholar]
- He H, Liyanarachchi S, Akagi K, et al. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science 2011 ; 332 : 238–240. [CrossRef] [PubMed] [Google Scholar]
- Padgett RA, Shukla GC. A revised model for U4atac/U6atac snRNA base pairing. RNA 2002 ; 8 : 125–128. [CrossRef] [PubMed] [Google Scholar]
- Liu S, Li P, Dybkov O, et al. Binding of the human Prp31 Nop domain to a composite RNA-protein platform in U4 snRNP. Science 2007 ; 316 : 115–120. [CrossRef] [PubMed] [Google Scholar]
- Pessa HKJ, Frilander MJ. Genetics. Minor splicing, disrupted. Science 2011 ; 332 : 184–185. [CrossRef] [PubMed] [Google Scholar]
© 2012 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1. Phénotype du MOPD1/TALS. A, B. Patient en période néonatale. A. On note un visage rond, un occiput proéminent, une protrusion oculaire, un nez busqué, un menton petit et en retrait, un cou court, une peau sèche, une absence de cheveux, des contractures articulaires des genoux et des coudes ainsi que des mains et pieds courts. B. Radiographies osseuses montrant, aux membres inférieurs, un retard d’ossification des épiphyses tibiales supérieures et fémorales inférieures (les flèches vertes indiquent la position attendue des épiphyses ossifiées, absentes chez le patient), des fémurs courts et incurvés et des métaphyses fémorales discrètement élargies (flèche rouge) et, au niveau de la colonne, un discret aplatissement des corps vertébraux (platyspondylie) et une cyphose lombaire (indiquée par le crochet blanc). C. Imagerie cérébrale par résonance magnétique (IRM) à l’âge de 1 mois. Kyste arachnoïdien (flèche blanche), hypoplasie cérébrale et pachygyrie (flèche jaune), agénésie de la partie antérieure du corps calleux (la pointe de la tête de flèche rouge montre la position attendue de la partie antérieure du corps calleux, ici absente) et discrète hypoplasie du vermis cérébelleux (flèche bleue). |
| Dans le texte | |
|
Figure 2. Modèle bidimensionnel de la bimolécule humaine formée des snARN U4atac/U6atac. La tige boucle située en 5’ du snRNA U4atac résulte d’un appariement intramoléculaire entre nucléotides complémentaires. Les mutations identifiées chez les patients MOPD1/TALS sont situées le plus souvent dans la tige boucle 5’ du snARN U4atac. Le site de fixation de la protéine 15.5K, indiqué par un disque bleu, est nécessaire à l’association avec le snARN U5 (adapté de [10] et [11]). |
| Dans le texte | |
|
Figure 3. Splicéosome mineur : une fonction vitale. Les défauts de l’épissage des introns de type U12 sont dus à un défaut de la fonction du splicéosome mineur (U12-dépendant). Les mutations MOPD1/TALS conduisent à une accumulation des transcrits prémessagers non épissés ; 700 à 800 gènes humains contenant des introns U12 ont probablement un niveau d’expression plus ou moins modifié (démontré dans notre étude [1] pour certains d’entre eux) (d’après [12]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.