")
")
| Issue |
Med Sci (Paris)
Volume 27, Number 10, Octobre 2011
|
|
|---|---|---|
| Page(s) | 814 - 816 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/20112710007 | |
| Published online | 21 October 2011 | |
Bases structurales du syndrome du choc toxique streptococcique
Structural basis for streptococcal toxic shock syndrome
1
Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, California 92093, États-Unis
2
Adresse actuelle : Biologie structurale des interactions entre virus et cellule hôte, UMI 3265, Université Joseph Fourier-EMBL-CNRS, 6, rue Jules Horowitz, 38042 Grenoble, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Streptococcus pyogenes, bactérie Gram positive, est un pathogène humain qui n’est en général responsable que d’infections mineures comme l’angine ou l’impétigo. Dans certains cas, il peut entraîner des affections plus sévères comme la fasciite nécrosante ou les rhumatismes articulaires aigus, voire la mort si un syndrome du choc toxique streptococcique (SCTS) survient [1]. Les premiers signes du SCTS sont la vasodilatation et l’altération de la barrière endothéliale sous l’effet de la production de la protéine HBP (heparin binding protein) par les polynucléaires neutrophiles. Une fuite importante de plasma sanguin en résulte qui entraîne la défaillance de plusieurs organes.
Le principal facteur de virulence de S. pyogenes est la protéine M, qui forme des fibrilles à la surface de la bactérie [2]. Elle est impliquée dans l’adhésion du pathogène aux cellules épithéliales, sa pénétration dans les tissus et sa protection contre la phagocytose [3, 4]. En outre, la protéine M permet à la bactérie d’échapper aux systèmes immunitaires inné et acquis. Parmi les différentes variantes génétiques de la protéine M, la plus virulente est le sérotype M1, qui a été identifié dans tous les types d’infection. Lors d’une infection par S. pyogenes, la production de HBP par les neutrophiles est déclenchée par la forte interaction entre la protéine M1 et le fibrinogène humain (Fg) qui conduit à l’agrégation de ces deux protéines. L’agrégat est reconnu par des récepteurs à la surface des neutrophiles, les intégrines β2, qui sont alors activés et déclenchent la sécrétion de HBP (Figure 1A) [5]. Afin de comprendre la première étape du mécanisme d’activation des neutrophiles et d’envisager le développement de nouveaux vaccins, nous avons entrepris une étude structurale du complexe entre les fragments de la protéine M1 et du fibrinogène humain qui interagissent [6].
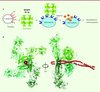 |
Figure 1. L’interaction entre M1 et le fibrinogène induit les symptômes du syndrome du choc toxique. A. La bactérie Streptococcus pyogenes sécrète une protéine de virulence dans la circulation sanguine, la protéine M1 (en rouge), grâce à l’action d’une protéase spécifique. M1 interagit alors dans le milieu avec le fibrinogène de l’hôte (en vert) pour former un agrégat amorphe qui sera par la suite reconnu par les récepteurs intégrines β2 à la surface des neutrophiles. Ces neutrophiles sont alors activés et sécrètent une protéine à fort potentiel vasodilatateur, l’heparin binding protein (HBP, en orange). B. Structure cristallographique du fragment M1BC1 (rouge) formant un complexe avec le fragment FgD du fibrinogène (en vert) qui permet de comprendre les bases de la formation de l’agrégat (adapté de [6]). |
Structure du complexe M1BC1-FgD
Nous avons résolu la structure cristallographique à 3,3 Å de résolution de ce complexe de grande taille (380 kDa). Il est composé du fragment M1BC1 de la protéine M1 et du fragment FgD du fibrinogène humain. M1BC1 contient notamment les motifs B1 et B2 responsables de l’accroche au fibrinogène. Le fragment D du fibrinogène comprend deux têtes globulaires responsables de l’interaction avec les intégrines β2. La structure du complexe présente globalement une organisation en croix avec quatre molécules de FgD entourant un dimère de M1BC1, lui-même organisé en double hélice (Figure 1B). Cela s’accorde bien avec la présence de 2 x 2 = 4 motifs B sur le dimère de M1. La répétition B1 fixe 2 molécules de FgD orientées à 180° ; il en est de même pour le motif B2. Comme B1 et B2 sont situés sur M1BC1 à un quart de tour d’hélice l’un de l’autre, les deux paires de FgD sont positionnées à angle droit et forment une croix.
Les agrégats de M1-Fg sont responsables de l’activation des neutrophiles
Pour comprendre le mécanisme d’activation des neutrophiles par l’agrégat M1-Fg, nous l’avons modélisé en utilisant notre structure du complexe M1BC1-FgD, la structure du fibrinogène Fg entier, et un modèle de la protéine M1 entière. Le modèle obtenu constitue un réseau tridimensionnel contenant des piliers de Fg articulés autour de nœuds de M1 (Figure 2A). Nous avons vérifié cette organisation par microscopie électronique (Figure 2B). Ce modèle nous permet de comprendre le mécanisme d’activation des neutrophiles. Il a déjà été montré que des anticorps capables de réticuler les récepteurs intégrines β2 avaient le même effet sur l’activation des neutrophiles que le complexe M1-Fg [5, 7] et que la formation du complexe M1-Fg devait induire la réticulation des intégrines en surface des neutrophiles. En partant de ce postulat, nous avons cherché à tester l’influence de la structure de l’agrégat M1-Fg sur l’activation des neutrophiles. Pour cela, nous avons préparé différents mutants de délétion de la protéine M1 et mesuré leur effet sur la sécrétion de HBP par les neutrophiles. Dans un premier temps, nous avons supprimé tour à tour une des deux répétitions B de M1, créant ainsi les mutants M1ΔB1 et M1ΔB2, et modélisé leur capacité à interagir avec le fibrinogène. Les deux mutants ont gardé leur aptitude à accrocher le fibrinogène in vitro mais forment une architecture bidimensionnelle en forme de fibre (Figure 2C, D). Des neutrophiles humains mis en présence de ces mutants n’induisent pas la sécrétion de HBP (Figure 2G), révélant que c’est la formation de l’agrégat, et non la capacité à accrocher le fibrinogène, qui est indispensable à l’activation des neutrophiles. Dans un deuxième temps, nous avons examiné l’influence de la densité de l’agrégat sur l’activation des neutrophiles. Pour cela, nous avons construit un mutant de M1 dont la répétition B2 amino-terminale a été supprimée puis réinsérée en position carboxy-terminale de la molécule (M1B2C) pour augmenter l’espace entre les deux répétitions B. La modélisation de l’agrégat modifié a confirmé son organisation moins dense que pour la protéine native (Figure 2E), différence que nous n’avons pas pu vérifier par microscopie électronique en raison de la résolution limitée (Figure 2F). Ce caillot plus lâche que le caillot natif n’induit pas non plus la sécrétion de HBP (Figure 2G), suggérant que la densité de l’agrégat est également cruciale pour l’activation des neutrophiles.
 |
Figure 2. La présence et la densité de l’agrégat déclenchent l’activation des neutrophiles. A. Modèle d’agrégat M1-Fg entre la protéine M1 (en rouge) et le fibrinogène humain (en vert) B. Cliché de microscopie électronique de l’agrégat M1-Fg natif. C. Modèle d’agrégat entre le fibrinogène humain et les mutants M1ΔB1 ou M1ΔB2. D. Cliché de microscopie électronique de l’agrégat correspondant. E. Modèle d’agrégat entre le fibrinogène humain et le mutant M1B2C. F. Cliché de microscopie électronique de l’agrégat correspondant. G. Visualisation par Western blot de la sécrétion de HBP par les neutrophiles mis en présence des différents mutants de M1, en utilisant des anticorps anti-HBP. La ligne rHBP correspond au contrôle positif de la protéine HBP recombinante (adapté de [6]). |
La réticulation des intégrines est responsable de l’activation des neutrophiles
Cette série d’expériences nous a permis de prouver qu’une des propriétés pro-inflammatoires de la protéine de virulence M1 de Streptococcus pyogenes consistait en son interaction avec le fibrinogène humain pour former un agrégat amorphe différent des caillots de fibrine fréquemment générés par le fibrinogène. Puisque la perturbation du réseau M1-fibrinogène en fibres ou en agrégats plus lâches conduit à la perte de l’activation des neutrophiles, nous avons conclu que la densité du fibrinogène dans le réseau est un aspect essentiel dans l’activation des neutrophiles. En effet, les agrégats de M1-fibrinogène forment un réseau avec les têtes globulaires de fibrinogène situées vers l’extérieur de l’assemblage supramoléculaire, ce qui permet l’accroche et la réticulation des intégrines β2. Ceci conforte l’hypothèse selon laquelle la concentration et la réticulation des intégrines sont des processus conservés dans les mécanismes d’activation des leucocytes. Il est ainsi possible d’imaginer que la désorganisation de l’interaction M1-fibrinogène puisse représenter une voie thérapeutique pour combattre les graves conséquences du syndrome du choc toxique streptococcique.à
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Références
- Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 2000 ; 13 : 470–511. [CrossRef] [PubMed] [Google Scholar]
- Ghosh P. The nonideal coiled coil of M protein and its multifarious functions in pathogenesis. Adv Exp Med Biol 2011 ; 715 : 197–211. [CrossRef] [PubMed] [Google Scholar]
- Fischetti VA. Streptococcal M protein: molecular design and biological behavior. Clin Microbiol Rev 1989 ; 2 : 285–314. [PubMed] [Google Scholar]
- McNamara C, Zinkernagel AS, Macheboeuf P, et al. Coiled-coil irregularities and instabilities in group A Streptococcus M1 are required for virulence. Science 2008 ; 319 : 1405–1408. [CrossRef] [PubMed] [Google Scholar]
- Herwald H, Cramer H, Mörgelin M, et al. M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell 2004 ; 116 : 367–379. [CrossRef] [PubMed] [Google Scholar]
- Macheboeuf P, Buffalo C, Fu CY, et al. Streptococcal M1 protein constructs a pathological host fibrinogen network. Nature 2011 ; 472 : 64–68. [CrossRef] [PubMed] [Google Scholar]
- Gautam N, Herwald H, Hedqvist P, et al. Signaling via beta2 integrins triggers neutrophil-dependent alteration in endothelial barrier function. J Exp Med 2000 ; 191 : 1829–1839. [CrossRef] [PubMed] [Google Scholar]
© 2011 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1. L’interaction entre M1 et le fibrinogène induit les symptômes du syndrome du choc toxique. A. La bactérie Streptococcus pyogenes sécrète une protéine de virulence dans la circulation sanguine, la protéine M1 (en rouge), grâce à l’action d’une protéase spécifique. M1 interagit alors dans le milieu avec le fibrinogène de l’hôte (en vert) pour former un agrégat amorphe qui sera par la suite reconnu par les récepteurs intégrines β2 à la surface des neutrophiles. Ces neutrophiles sont alors activés et sécrètent une protéine à fort potentiel vasodilatateur, l’heparin binding protein (HBP, en orange). B. Structure cristallographique du fragment M1BC1 (rouge) formant un complexe avec le fragment FgD du fibrinogène (en vert) qui permet de comprendre les bases de la formation de l’agrégat (adapté de [6]). |
| Dans le texte | |
|
Figure 2. La présence et la densité de l’agrégat déclenchent l’activation des neutrophiles. A. Modèle d’agrégat M1-Fg entre la protéine M1 (en rouge) et le fibrinogène humain (en vert) B. Cliché de microscopie électronique de l’agrégat M1-Fg natif. C. Modèle d’agrégat entre le fibrinogène humain et les mutants M1ΔB1 ou M1ΔB2. D. Cliché de microscopie électronique de l’agrégat correspondant. E. Modèle d’agrégat entre le fibrinogène humain et le mutant M1B2C. F. Cliché de microscopie électronique de l’agrégat correspondant. G. Visualisation par Western blot de la sécrétion de HBP par les neutrophiles mis en présence des différents mutants de M1, en utilisant des anticorps anti-HBP. La ligne rHBP correspond au contrôle positif de la protéine HBP recombinante (adapté de [6]). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.