")
")
| Issue |
Med Sci (Paris)
Volume 27, Number 6-7, Juin–Juillet 2011
|
|
|---|---|---|
| Page(s) | 579 - 581 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2011276006 | |
| Published online | 01 July 2011 | |
Prédisposition mendélienne aux infections mycobactériennes et défaut de l’explosion oxydative des macrophages
Mendelian susceptibility to mycobacterial infections and defect in macrophages respiratory burst
Laboratoire de génétique humaine des maladies infectieuses, Inserm U980 ; Université Paris Descartes, Faculté de médecine Necker, 156, rue de Vaugirard, 75015 Paris, France
*
This email address is being protected from spambots. You need JavaScript enabled to view it.
Étiologie génétique du syndrome de prédisposition mendélienne aux infections mycobactériennes
La tuberculose est une maladie qui est encore actuellement un important problème de santé publique. L’étude de la prédisposition génétique à la tuberculose est un domaine de recherche très riche et actif [1]. Le syndrome de prédisposition mendélienne aux infections mycobactériennes (MSMD, Mendelian susceptibility to mycobacterial disease - MIM 209950) est un syndrome rare caractérisé par une susceptibilité anormale à des germes intracellulaires peu virulents (comme le bacille vaccinal de Calmette-Guérin et les mycobactéries environnementales) sans présence évidente d’une autre infection (virus, bactéries, champignons, parasites) [2]. Ces patients peuvent également présenter des infections à Mycobacterium tuberculosis [3]. Différentes mutations causales ont été décrites dans six gènes autosomaux : IFNGR1 et IFNGR2 qui codent pour des interferon gamma receptors, STAT1 qui code pour le signal transducer and activator of transcription 1, IL12B et IL12RB1 qui codent pour l’interleukine 12 et la chaîne bêta 1 du récepteur de l’interleukine 12, interferon regulator factor 8 (IRF8) et un gène situé sur le chromosome X, nuclear factor-kB essential modulator (NEMO). Ces mutations affectent la boucle d’activation existant entre l’IL-12, l’IL-23 et l’IFNγ (interféron γ) [4, 5] (Figure 1). L’hétérogénéité allélique de ce syndrome est liée en partie à la diversité des anomalies moléculaires responsables. Toutefois, l’étiologie génétique reste encore inconnue chez la moitié des patients présentant un MSMD.
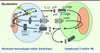 |
Figure 1 Étiologies génétiques du syndrome de prédisposition mendélienne aux infections mycobactériennes. L’IL-12 est sécrétée par les monocytes, macrophages et cellules dendritiques et se lie à son récepteur (composé de deux chaînes, β1et β2) exprimé sur les lymphocytes T et les cellules NK (natural killer). L’IFN-γ est sécrété par les lymphocytes T et les cellules NK et se lie à son récepteur ubiquitaire composé de deux chaînes (IFN-γR1 et IFN-γR2). STAT-1 est phosphorylé en réponse à l’IFN-γ et migre dans le noyau sous forme d’hétérodimères. NEMO est une protéine régulatrice de la voie NF-қB. La signalisation CD40 de la voie NF-қB est importante pour la production d’IL-12 et pour l’immunité antimycobactérienne. IRF8 est un facteur de transcription induit par l’IFN-γ et nécessaire à l’induction des gènes cibles comme l’IL-12. Gp91phox est le composant majeur de la NADPH oxydase. Les molécules mutées chez les patients MSMD sont indiquées en gris. L’hétérogénéité allélique de ces huit gènes définit au moins 15 maladies génétiques différentes. Les mutations Q231P et T178P gp91phox affectent spécifiquement l’explosion oxydative des macrophages dérivés in vitro; il est probable que gp91phox induise la libération d’IL-12 (ceci n’est pas encore démontré). |
Nous avons étudié deux familles françaises chez lesquelles sept hommes apparentés par leurs mères présentaient une infection mycobactérienne, suggérant une transmission récessive liée à l’X [6]. Ces patients ont développé des infections, pour six d’entre eux à Mycobacterium bovis-BCG (BCG-ite : infection locale ou régionale ; ou BCG-ose : infection disséminée) et pour un autre patient à Mycobacterium tuberculosis. Une femme, conductrice obligatoire, avait également présenté une salpingite tuberculeuse. Toutes les causes de déficits immunitaires primaires liés au chromosome X avaient été éliminées par des études fonctionnelles et par une caractérisation clinique fine [6]. Aucun défaut de l’axe IL-12/IL-23/IFN-γ n’a été mis en évidence par les tests fonctionnels utilisés pour le diagnostic des étiologies génétiques connues du MSMD. Par une analyse génétique de liaison sur le chromosome X, nous avons identifié deux régions, avec un LOD score (logarithm of the odds ratio) de 2,29 après une analyse multipoints en Xp11.3-Xp21.1 et en Xq25-Xq26.3 [7]. De façon inattendue, en amplifiant le gène CYBB, nous avons trouvé une mutation germinale correspondant au remplacement d’une base A par C qui entraînait un changement de l’acide aminé (aa) 231 (glutamine [Q] remplacé par proline [P], Q231P), chez les quatre hommes malades dans une famille et à l’état hémizygote, et un changement de l’aa 178 (thréonine [T] par proline [P], T178P) chez les trois hommes malades de l’autre famille. En plus, il existait une coségrégation familiale entre la mutation de CYBB et le phénotype infectieux mycobactérien [7].
CYBB, un nouveau gène de prédisposition aux MSMD lié à l’X
CYBB et granulomatose septique chronique liée à l’X
CYBB est un gène codant la protéine gp91phox, qui est essentielle à la NADPH (nicotinamide adénine dinucléotide phosphate) oxydase, responsable de l’explosion oxydative dans les cellules phagocytaires (neutrophiles, monocytes, macrophages). La NADPH oxydase induit une réduction de l’oxygène moléculaire en anion superoxyde (O2 -) à l’origine d’autres formes réactives de l’oxygène [13]. Les réactifs dérivés de l’oxygène (ROS) sont produits par la dismutation du superoxyde [14]. Les phagocytes sont capables de produire directement de l’ion superoxyde (O2-), une molécule instable qui sera ensuite transformée en peroxyde d’hydrogène (H2O2) sous l’action de la superoxyde dismutase. La NADPH oxydase est constituée de la flavocytochrome b558 membranaire qui est un hétérodimère formé par une molécule de gp91phox et par une molécule de p22phox, de trois éléments cytoplasmiques (p47phox, p67phox et p40phox) et de petites protéines GTPases. Les mutations de CYBB sont responsables de la forme liée à l’X de la granulomatose septique chronique (CGD pour chronic granulomatose disease) [8]. Cette maladie est un déficit immunitaire primaire lié à une anomalie de la NADPH oxydase. Des mutations dans NCF1, NCF2, NCF4 (codant les neutrophil cytosolic factors 1, 2, 4) et CYBA permettent de définir les formes autosomiques récessives de la CGD [9]. Les patients atteints de cette maladie présentent des infections bactériennes et fongiques récurrentes et/ou disséminées et ont, sur le plan biologique, un défaut d’explosion oxydative dans l’ensemble de leurs cellules phagocytaires. En général, l’activité nulle ou fortement réduite de la NADPH oxydase est mise en évidence par des techniques utilisant le NBT (nitrobleu de tétrazolium) ou le DHR (réduction de la dihydrorhodamine) ou la réduction du cytochrome-c [10]. Ces patients présentent rarement des infections mycobactériennes [11, 12]. Chez tous ces patients existe un défaut de l’explosion oxydative dans l’ensemble des cellules phagocytaires.
Particularités des mutations Q231P-CYBB et T178P-CYBB responsables du MSMD
Les mutations Q231P-CYBB et T178P-CYBB que nous avons découvertes étaient particulièrement intéressantes parce qu’elles n’avaient jamais été décrites. Les tests effectués sur les granulocytes à la recherche du diagnostic de CGD étaient normaux alors que les patients avaient un phénotype clinique d’infection mycobactérienne. Or il existait une dissociation des phénotypes cellulaires chez les patients porteurs de ces deux mutations identifiées Q231P-CYBB et T178P-CYBB. Les études effectuées sur l’ensemble des cellules phagocytaires étaient homogènes : la production de O2-, de H2O2, et la libération de H2O2 montraient une activité de la NADPH oxydase normale et comparable à celle des polynucléaires neutrophiles et des monocytes (cellules circulantes) de sujets normaux (Figure 2A, B) et la capacité de tuer le Staphylococcus aureus était également normale [7]. En revanche, il n’y avait ni activité NADPH oxydase ni explosion oxydative dans les macrophages dérivés in vitro (Figure 2C). L’explosion oxydative dans les lignées dérivées de lymphocytes B immortalisés par le virus d’Epstein Barr (lignées B-EBV) de sept patients était également abolie. De plus, l’expression de la protéine gp91phox mesurée dans les extraits protéiques de macrophages dérivés in vitro et de lignées B-EBV était nettement réduite. L’impact spécifique de cette mutation sur les macrophages était corrélé avec l’expression protéique de gp91phox et avec l’assemblage de la flavocytochrome b558 [7]. Les études de transfection stable des mutants Q231P-CYBB et T178P-CYBB réalisées sur les lignées CHO et les lignées PLB-985 corrèlent avec les mesures d’expression protéique obtenues à partir des cellules des patients [7]. Les deux mutations de CYBB conduisent donc à l’abolition de l’expression de la protéine dans deux types cellulaires (les macrophages dérivés in vitro et les lignées B-EBV) alors qu’elle est normale dans deux autres types cellulaires (granulocytes et monocytes). Il s’agit du premier exemple d’une maladie génétique mimant le déficit créé chez des souris par les approches de knock-out conditionnel [7].
 |
Figure 2 Altérations de l’activité NADPH des cellules phagocytaires porteuses de mutations du gène CYBB. Quantification fluorimétrique de la libération de H2O2 dans les polynucléaires neutrophiles (PMN) (A), les monocytes (B), les macrophages (C) dérivés in vitro de contrôles sains (C+), de patients atteints d’une granulomatose septique chronique (C-), et de patients ayant une mutations de CYBB (Q321P et T178P). Les cellules A et B ont été activées ou non avec un ester de phorbol, PMA (400 ng/ml); en (C), une deuxième activation a été effectuée utilisant l’IFN-γ; le substrat fluorogénique est ADHP (N-acétyl-3,7 dihydrophénoxazine). |
Ce travail a donc permis d’identifier une deuxième forme génétique du MSMD liée à l’X, d’identifier une nouvelle forme génétique du syndrome de prédisposition mendélienne à la tuberculose, de démontrer que MSMD et CGD peuvent être alléliques au locus CYBB, de démontrer que l’explosion oxydative dans les macrophages joue un rôle essentiel pour le contrôle de l’infection mycobactérienne. Les autres infections retrouvées communément dans la CGD sont vraisemblablement dues à un défaut de la fonction oxydative dans les granulocytes et les monocytes. Cette étude permet d’offrir à l’ensemble des patients un diagnostic moléculaire permettant un conseil génétique et des perspectives de traitements fondés sur la physiopathologie. Il reste à démontrer comment s’établit la connexion entre l’axe IL-12-IL-23/IFNγ et le système NADPH oxydase. Ce travail offre aussi des perspectives de recherche très nouvelles pour l’étude de la prédisposition génétique aux infections mycobactériennes disséminées et/ou récurrentes, comme la tuberculose.
Conflit d’intérêts
L’auteur déclare n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Références
- Alcais A, Fieschi C, Abel L, Casanova JL. Tuberculosis in children and adults: two distinct genetic diseases. J Exp Med 2005 ; 202 : 1617-1621. [CrossRef] [PubMed] [Google Scholar]
- Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol 2002 ; 20 : 581-620. [CrossRef] [PubMed] [Google Scholar]
- Boisson-Dupuis S, El Baghdadi J, Parvaneh N, et al. IL-12RB1 deficiency in two of fifty children with severe tuberculosis from Iran, Morocco, Turkey. PLoS One. PLoS One 2011 ; 6 : e18524. [CrossRef] [PubMed] [Google Scholar]
- Filipe-Santos O, Bustamante J, Chapgier A, et al. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol 2006 ; 18 : 347-361. [CrossRef] [PubMed] [Google Scholar]
- Hambleton S, Salem S, Bustamante J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med 2011 ; 27 : avril, online. [Google Scholar]
- Bustamante J, Picard C. A novel X-linked recessive form of Mendelian susceptibility to mycobaterial disease. J Med Genet 44 : e65. [Google Scholar]
- Bustamante J, Arias AA, Vogt G, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol 2011 ; 12 : 213-221. [CrossRef] [PubMed] [Google Scholar]
- Roos D, Kuhns DB, Maddalena A, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis 2010 ; 45 : 246-265. [CrossRef] [PubMed] [Google Scholar]
- Roos D, Kuhns DB, Maddalena A, et al. Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol Dis 2010 ; 44 : 291-299. [CrossRef] [PubMed] [Google Scholar]
- Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med 2010 ; 363 : 2600-2610. [CrossRef] [PubMed] [Google Scholar]
- Winkelstein JA, Marino MC, RB Johnston, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 2000 ; 79 : 155-169. [CrossRef] [PubMed] [Google Scholar]
- Bustamante J, Aksu G, Vogt G, et al. BCG-osis and tuberculosis in a child with chronic granulomatous disease. J Allergy Clin Immunol 2007 ; 120 : 32-38. [CrossRef] [PubMed] [Google Scholar]
- Gougerot-Pocidalo MA, El Benna J, My-Chan Dang P, Elbim C. Quand les polynucléaires neutrophiles attrapent les agents pathogènes dans leurs filets. Med Sci (Paris) 2007 ; 23 : 464-465. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Migdal C, Serres M. Espèces réactives de l’oxygène et stress oxydant. Med Sci (Paris) 2011 ; 27 : 405-412. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
© 2011 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1 Étiologies génétiques du syndrome de prédisposition mendélienne aux infections mycobactériennes. L’IL-12 est sécrétée par les monocytes, macrophages et cellules dendritiques et se lie à son récepteur (composé de deux chaînes, β1et β2) exprimé sur les lymphocytes T et les cellules NK (natural killer). L’IFN-γ est sécrété par les lymphocytes T et les cellules NK et se lie à son récepteur ubiquitaire composé de deux chaînes (IFN-γR1 et IFN-γR2). STAT-1 est phosphorylé en réponse à l’IFN-γ et migre dans le noyau sous forme d’hétérodimères. NEMO est une protéine régulatrice de la voie NF-қB. La signalisation CD40 de la voie NF-қB est importante pour la production d’IL-12 et pour l’immunité antimycobactérienne. IRF8 est un facteur de transcription induit par l’IFN-γ et nécessaire à l’induction des gènes cibles comme l’IL-12. Gp91phox est le composant majeur de la NADPH oxydase. Les molécules mutées chez les patients MSMD sont indiquées en gris. L’hétérogénéité allélique de ces huit gènes définit au moins 15 maladies génétiques différentes. Les mutations Q231P et T178P gp91phox affectent spécifiquement l’explosion oxydative des macrophages dérivés in vitro; il est probable que gp91phox induise la libération d’IL-12 (ceci n’est pas encore démontré). |
| Dans le texte | |
|
Figure 2 Altérations de l’activité NADPH des cellules phagocytaires porteuses de mutations du gène CYBB. Quantification fluorimétrique de la libération de H2O2 dans les polynucléaires neutrophiles (PMN) (A), les monocytes (B), les macrophages (C) dérivés in vitro de contrôles sains (C+), de patients atteints d’une granulomatose septique chronique (C-), et de patients ayant une mutations de CYBB (Q321P et T178P). Les cellules A et B ont été activées ou non avec un ester de phorbol, PMA (400 ng/ml); en (C), une deuxième activation a été effectuée utilisant l’IFN-γ; le substrat fluorogénique est ADHP (N-acétyl-3,7 dihydrophénoxazine). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.