")
")
| Issue |
Med Sci (Paris)
Volume 27, Number 5, Mai 2011
|
|
|---|---|---|
| Page(s) | 473 - 475 | |
| Section | Nouvelles | |
| DOI | https://doi.org/10.1051/medsci/2011275009 | |
| Published online | 25 May 2011 | |
Les β-thalassémies
Espoirs thérapeutiques de l’hepcidine
Thalassemia
Therapeutic hopes carried by hepcidin
1
Inserm U1016, CNRS UMR 8104, Institut Cochin, 24, rue du Faubourg Saint-Jacques, 75014 Paris, France
2
Inserm U567, CNRS UMR 8104, Institut Cochin, 24, rue du Faubourg Saint-Jacques, 75014 Paris, France
L’homéostasie du fer dans l’organisme repose sur un contrôle strict de l’absorption du fer au niveau des entérocytes matures de la villosité duodénale. L’hepcidine, une petite protéine synthétisée par l’hépatocyte, s’est révélée être un peptide hormonal clé du métabolisme du fer capable d’inhiber l’absorption intestinale du fer alimentaire.
Il y a deux ans, nous rapportions l’identification d’un facteur érythroïde, GDF15 - membre de la superfamillle TGFβ (transforming growth factor beta) -, dont la production médullaire est très augmentée chez les patients β-thalassémiques. Cela pouvait entraîner l’inhibition de la synthèse d’hepcidine et donc, indirectement, faciliter la surcharge en fer caractéristique de l’érythropoïèse inefficace chez ces patients [1]. Si peu de progrès ont été faits sur les mécanismes moléculaires de la répression de l’hepcidine, en revanche, un pas considérable dans la démonstration de l’effet délétère de cette répression de l’hormone sur la surcharge en fer et les anomalies de l’érythropoïèse associées à la thalassémie a été franchi. Dans un article du Journal of clinical investigation, Gardenghi et al. apportent la démonstration des effets thérapeutiques de l’hepcidine dans la thalassémie et montrent que cette hormone agit à un double niveau en corrigeant la surcharge en fer et en améliorant les paramètres hématologiques [2].
Les thalassémies
Histoire naturelle et défauts moléculaires
Les β-thalassémies sont des maladies génétiques parmi les plus fréquentes ; elles sont dues à un déficit plus ou moins complet de synthèse de la chaîne β de l’hémoglobine (Hb). La forme la plus sévère est l’anémie de Cooley (bêta-thalassémie homozygote majeure). Les sujets sont profondément anémiques (Hb ≤ 7 g/dl), anémie que potentialise l’excès relatif de chaînes α-globine qui précipitent, entraînant lésions membranaires et destruction des érythroblastes, formation d’hémichromes et production de radicaux oxydatifs libres (ROS). Dans ces cas de β-thalassémie sévère, une érythropoïèse inefficace est constante, avec déformations osseuses caractéristiques (par expansion du volume médullaire), hépatomégalie et splénomégalie importantes (Figure 1). Dans l’histoire naturelle de la maladie, la survie des enfants ne dépassait souvent pas la première année, rarement l’âge de 8-10 ans. Des formes moins sévères existent, définies comme thalassémies intermédiaires, qui ne requièrent pas de transfusions, ou seulement de façon épisodique (taux d’Hb de 7 à 10 g/dl). Les thalassémies ont une incidence accrue dans certaines parties du monde comme le bassin méditerranéen, d’où le nom de thalassémie (θαλασα = la mer), mais à une plus grande échelle l’Inde, l’Asie du Sud-Est, le sud de la Chine où elles peuvent représenter un problème de santé majeur.
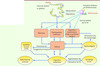 |
Figure 1 Conséquences de la production excessive de chaînes α globine dans la β-thalassémie. Les processus primaires sont indiqués en orange et les processus compensatoires en jaune.EPO : érythropoïétine; GR : globules rouges. D’après [11]. |
Les défauts moléculaires sont majoritairement des mutations ponctuelles ou des microdélétions/additions touchant toutes les régions codantes ou non codantes du gène globine, et entravant toutes les étapes de la biosynthèse de la protéine. On distingue β0-thalassémies et β+-thalassémies, selon qu’il y a, ou non, synthèse partielle d’Hb, mais aucune relation génotype/phénotype constante n’a pu être établie. Une caractéristique des thalassémies, conséquence de l’érythopoïèse inefficace, est une absorption accrue de fer au niveau des entérocytes, avec saturation progressive de la transferrine. Associée aux transfusions sanguines, elle explique que la surcharge en fer soit constante dans la bêta-thalassémie homozygote.
Approche thérapeutique
La première approche thérapeutique, corrigeant l’anémie par transfusion, a profondément modifié l’évolution de la maladie et permis la survie des patients. Cependant, il faut lui associer en continu un traitement chélateur du fer puisqu’elle entraîne une accumulation massive pathologique du fer, considérée comme la source principale des complications de la β-thalassémie. Dès la deuxième ou troisième décennie de la vie, la surcharge en fer domine le tableau clinique avec des troubles systémiques : dépôts au niveau du myocarde et des glandes endocrines, diabète, troubles hépatiques, et les sujets meurent de défaillance cardiaque entre 20 et 30 ans. La desferoxamine injectable est le principal chélateur, auquel s’ajoutent des chélateurs oraux, défériprone et déférasirox [3]. Le seul traitement curateur de la thalassémie est la greffe de moelle osseuse à partir d’un donneur HLA compatible qui a donné d’excellents résultats en Italie, puis en France, sur des séries de quelques centaines de patients [4]. L’avenir de la thérapie génique, malgré un succès récent dans un cas prototype [5], n’est pas connu, et cette approche semblerait pour l’instant inadaptée aux besoins économiques et démographiques.
C’est dans cette perspective qu’un abord thérapeutique par l’hepcidine a été envisagé dans un modèle murin de thalassémie. La souris th3/+ utilisée est hétérozygote pour une délétion qui élimine les deux gènes βmajeur et βmineur, et présente un ensemble de symptômes comparable à celui d’une thalassémie intermédiaire, avec anémie, cellules érythroïdes immatures circulantes, accumulation de fer splénique et hépatique, élévation du fer sérique et de la saturation de la transferrine. Malheureusement, les souris th3/th3 chez lesquelles les gènes β-globine sont absents meurent au stade embryonnaire.
L’hepcidine, un outil thérapeutique pour les thalassémies
L’hepcidine, l’hormone du fer
L’hepcidine est un petit peptide hormonal (25 acides aminés) produit essentiellement par le foie et déversé dans la circulation. Ce peptide constitue l’hormone centrale de l’équilibre du fer dans l’organisme. L’hepcidine empêche l’export du fer des entérocytes, site de l’absorption intestinale du fer alimentaire, et des macrophages, site de recyclage du fer de l’hémoglobine, en dégradant la ferroportine, exportateur du fer présent à la membrane de ces cellules [6]. L’hepcidine agit donc comme une hormone hyposidérémiante.
La production d’hepcidine est augmentée par le fer, permettant ainsi de limiter l’accumulation du métal responsable de lésions tissulaires irréversibles et favorisant la production de ROS. À l’inverse, dans les conditions où l’organisme a besoin de fer (apport insuffisant, érythropoïèse accrue, hypoxie, etc.) la synthèse d’hepcidine est réprimée pour permettre une mobilisation efficace du métal. Les signaux contrôlant cette réponse sont probablement multiples : diminution de la saturation de la transferrine, taux d’érythropoïétine (dont l’injection provoque une répression de l’expression de l’hormone), facteurs solubles érythropoïétiques non encore identifiés. L’hepcidine se présente donc comme un « ferrostat » qui permet d’ajuster les quantités de fer aux demandes de l’organisme [7].
De nombreuses pathologies sont directement associées à la dérégulation de la production de l’hormone : d’une part, les maladies de surcharge en fer primaires (hémochromatoses), associées à un défaut de production d’hepcidine, et d’autre part, les situations d’hyperhepcidinémie associées aux anémies des maladies chroniques, ou bien encore les IRIDA (iron-refractory iron deficiency anemia), une forme génétique d’anémie induite par des mutations de la matriptase 2 [8]. L’identification des protéines impliquées dans ces maladies a permis une avancée importante dans la compréhension des mécanismes moléculaires de la régulation physiopathologique de l’hepcidine.
Chez les patients thalassémiques l’anémie s’accompagne d’une surcharge en fer secondaire, due à l’hyperabsorption intestinale et aggravée par le traitement transfusionnel. Plusieurs études ces dernières années ont montré que, probablement en réponse à l’augmentation de l’érythropoïèse, les niveaux d’hepcidine étaient très bas chez les patients thalassémiques, et que cette diminution du taux de l’hormone était probablement responsable de l’hyperabsorption intestinale de fer [9].
Effets thérapeutiques de l’hepcidine
Dans le travail présenté par Gardenghi et al., les auteurs partent d’une hypothèse simple : la surcharge en fer des souris β-thalassémiques pourrait être évitée en limitant chez ces souris la quantité de fer absorbée [2].
Pour tester cette hypothèse, les auteurs soumettent, dans une première série d’expériences, les souris β-thalassémiques à un régime pauvre en fer et montrent qu’effectivement elles accumulent moins de fer dans les tissus. De façon plus inattendue mais non moins intéressante, ils constatent qu’alors même que l’érythropoïèse n’est que peu affectée, la splénomégalie et l’anémie sont très sensiblement améliorées par ce régime. De ces observations, ils concluent que les souris β-thalassémiques absorbent plus de fer que ce qui est nécessaire pour leur érythropoïèse. Cette hyper-absorption de fer étant corrélée à des niveaux bas d’hepcidine, la deuxième série d’expériences a consisté à faire remonter les taux d’hepcidine chez les souris β-thalassémiques. Pour cela les souris β-thalassémiques ont été croisées avec des souris transgéniques exprimant de façon constitutive l’hepcidine. Chez les souris β-thalassémiques exprimant le transgène codant l’hepcidine, l’anémie s’améliore et on observe une diminution de la teneur en fer des tissus à l’âge de 1 et 5 mois. De façon très intéressante, les auteurs montrent des niveaux réduits de chaînes α-globine associées à la membrane et une diminution des niveaux de ROS dans les globules rouges : cela est compatible avec l’idée que, in fine, l’augmentation d’hepcidine a permis d’améliorer la cause primaire des dommages cellulaires et la mort prématurée des globules rouges. Il reste cependant à définir si cette amélioration résulte d’une diminution de l’érythrophagocytose macrophagique, d’une augmentation de la durée de vie des globules rouges, ou des deux.
De façon importante, la correction phénotypique observée semble dépendante des niveaux d’hepcidine apportés par le transgène. En effet, les auteurs constatent que des souris doubles transgéniques exprimant des niveaux élevés d’hepcidine non seulement ne sont pas corrigées mais aggravent leur anémie, suggérant une étroite fenêtre thérapeutique pour l’activité de l’hepcidine dans ce modèle (Figure 2).
 |
Figure 2 Effets de l’hepcidine sur les anomalies physiopathologiques de la β-thalassémie. Chez la souris thalassémique, les niveaux d’hepcidine sont bas, l’absorption du fer et le fer tissulaire sont augmentés et l’érythropoïèse inefficace conduit à l’anémie. L’addition d’hepcidine par transgenèse permet, lorsque les taux d’hepcidine sont revenus à la normale, d’améliorer les paramètres hématologiques et le statut martial des souris (surlignage rose). En revanche, les effets thérapeutiques de l’hepcidine sont perdus lorsque les concentrations de l’hepcidine deviennent supraphysiologiques (dernière ligne). |
Enfin, les données de Gardenghi et al. permettent de mieux comprendre les résultats récents de Li et al. [10] montrant que lorsque des souris thalassémiques recoivent des injections répétées de transferrine, la protéine de transport du fer sérique essentielle pour l’érythropoïèse, leur phénotype s’améliore et que des constatations très similaires à celles du travail de Gardenghi et al. sont faites, avec notamment une augmentation de la production d’hepcidine. Il est probable que dans ce modèle également, ce sont les effets sur les globules rouges en aval de l’hepcidine qui sont bénéfiques.
En conclusion, les données de Gardenghi et al. et de Li et al. sont très encourageantes et laissent penser que toute thérapeutique visant à augmenter les niveaux d’hepcidine, ou utilisant des agonistes de l’hormone, pourrait être bénéfique à un double niveau chez les patients thalassémiques, par correction de la surcharge en fer et amélioration de l’anémie.
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Références
- Vaulont S, Labie D. GDF15 coupable de l’hypersidérémie des patients thalassémiques par extinction de l’hepcidine. Med Sci (Paris) 2008 ; 24 : 139-141. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Gardenghi S, Ramos P, Marongiu MF, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest 2010 ; 120 : 4466-4477. [CrossRef] [PubMed] [Google Scholar]
- Hershko C. Pathogenesis and management of iron toxicity in thalassemia. Ann NY Acad Sci 2010 ; 1202 : 1-9. [CrossRef] [Google Scholar]
- Isgro A, Gaziev J, Sodani P, Lucarelli G. Progress in hematopoietic stem cell transplantation as allogeneic cellular gene therapy in thalassemia. Ann NY Acad Sci 2010 ; 1202 : 149-154. [CrossRef] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010 ; 16 : 318-322. [CrossRef] [PubMed] [Google Scholar]
- Viatte L, Vaulont S. Hepcidin, the iron watcher. Biochimie 2009 ; 91 : 1223-1228. [CrossRef] [PubMed] [Google Scholar]
- Lee PL, Beutler E. Regulation of hepcidin and iron-overload disease. Annu Rev Pathol 2009 ; 4 : 489-515. [CrossRef] [PubMed] [Google Scholar]
- Ganz T, Nemeth E. Hepcidin and disorders of iron metabolism. Annu Rev Med 2011 ; 62 : 347-360. [CrossRef] [PubMed] [Google Scholar]
- Nemeth E. Hepcidin in beta-thalassemia. Ann NY Acad Sci 2010 ; 1202 : 31-35. [CrossRef] [Google Scholar]
- Li H, Rybicki AC, Suzuka SM, et al. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med 2010 ; 16 : 177-182. [CrossRef] [PubMed] [Google Scholar]
- Olivieri NF. The beta-thalassemias. N Engl J Med 1999 ; 341 : 99-109. [CrossRef] [PubMed] [Google Scholar]
© 2011 médecine/sciences – Inserm / SRMS
Liste des figures
|
Figure 1 Conséquences de la production excessive de chaînes α globine dans la β-thalassémie. Les processus primaires sont indiqués en orange et les processus compensatoires en jaune.EPO : érythropoïétine; GR : globules rouges. D’après [11]. |
| Dans le texte | |
|
Figure 2 Effets de l’hepcidine sur les anomalies physiopathologiques de la β-thalassémie. Chez la souris thalassémique, les niveaux d’hepcidine sont bas, l’absorption du fer et le fer tissulaire sont augmentés et l’érythropoïèse inefficace conduit à l’anémie. L’addition d’hepcidine par transgenèse permet, lorsque les taux d’hepcidine sont revenus à la normale, d’améliorer les paramètres hématologiques et le statut martial des souris (surlignage rose). En revanche, les effets thérapeutiques de l’hepcidine sont perdus lorsque les concentrations de l’hepcidine deviennent supraphysiologiques (dernière ligne). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.