")
")
| Issue |
Med Sci (Paris)
Volume 26, Number 10, Octobre 2010
|
|
|---|---|---|
| Page(s) | 836 - 841 | |
| Section | M/S revues | |
| DOI | https://doi.org/10.1051/medsci/20102610836 | |
| Published online | 15 octobre 2010 | |
De la levure aux maladies neurodégénératives
Dix ans d’exploration des pathologies de la dynamique mitochondriale
From yeast to neurodegenerative diseases: ten years of exploration of mitochondrial dynamic disorders
1 Centre hospitalier universitaire d’Angers, 49933 Angers Cedex 9, France
2 Inserm U583, Institut des neurosciences de Montpellier, Hôpital Saint-Éloi, 80, rue Augustin Fliche, 34091 Montpellier Cedex 5, France
*
Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
L’identification, il y a dix ans, d’altérations de la dynamique mitochondriale dans des maladies neurodégénératives héréditaires affectant principalement les nerfs optiques et périphériques a signé l’émergence d’un nouveau type de mécanisme physiopathologique responsable de mitochondriopathies. Celui-ci implique des mutations dans les gènes codant des GTPases de la famille des dynamines qui assurent la fusion ou la fission du réseau mitochondrial. Des altérations similaires de la dynamique mitochondriale ont aussi été récemment retrouvées dans les maladies d’Alzheimer et de Parkinson, soulignant le rôle essentiel joué par la plasticité mitochondriale dans la survie neuronale, par le contrôle de l’activité énergétique et de sa distribution le long des axones.
Abstract
Ten years ago, OPA1 was identified as the major gene responsible for hereditary optic nerve degeneration, evidencing the first defect in mitochondrial network dynamics as the princeps pathophysiological mechanism in a mitochondriopathy. Later, alterations in other genes involved in mitochondrial fusion or fission, such as MFN2, DRP1 and GDAP1, were also associated with inherited neurological diseases, mainly affecting peripheral nerves. More recently, altered mitochondrial plasticity was also demonstrated in common age-related neurodegenerative disorders, as Alzheimer and Parkinson diseases, thus substantiating the critical role of mitochondrial dynamics in neurons as a key element governing the efficiency of oxidative respiration and its distribution along the axons.
© 2010 médecine/sciences - Inserm / SRMS
Il y a 10 ans, en 2000, dans le cadre d’études s’intéressant aux processus de fusion et de fission des mitochondries, nous avons identifié in silico l’orthologue humain des gènes MGM1 et Msp1 de Saccharomyces cerevisiae et Schizosaccharomyces pombe, impliqués dans les mécanismes de fusion des mitochondries et de maintien de leur génome. Ce gène, codant une GTPase mitochondriale de la famille des dynamines, est localisé sur le chromosome 3 au niveau du locus morbide OPA1 (optic atrophy 1), génétiquement lié à l’atrophie optique autosomique dominante, ou maladie de Kjer (AOAD, MIM 165500). En collaboration avec des équipes d’ophtalmo-généticiens, nous avons réalisé le séquençage de ce nouveau gène chez six familles atteintes de cette pathologie ; celui-ci révèla la présence systématique d’une mutation pathogène. L’orthologue de MGM1/Msp1 était donc bien le gène OPA1 [1]. Parallèlement, par une approche classique de clonage positionnel, une équipe germano-britannique arriva à la même conclusion [2]. Ces deux articles, publiés dans Nature Genetics en 2000, marquèrent le début de l’exploration des pathologies impliquant les processus de fusion/fission mitochondriale. Deux autres gènes codant des dynamines mitochondriales (MFN2, mitofusine 2 et DRP1, dynamin-related protein 1) ont ensuite été impliqués dans une forme axonale de la maladie de Charcot-Marie-Tooth (CMT) [3, 36] (→) et un syndrome neurologique néonatal sévère [4]. Enfin, un quatrième gène codant la protéine GDAP1 (ganglioside-induced differentiation-associated protein 1) impliquée dans le contrôle de la fission mitochondriale, fut associé à deux autres formes de CMT [5, 6]. Ces pathologies neurologiques, qui semblent liées à une altération de la plasticité mitochondriale, définissent une nouvelle classe de maladies mitochondriales, en marge de celles dues à un déficit primitif de la chaîne respiratoire. Les mécanismes physiopathologiques mis en jeu sont fort complexes puisque la plasticité mitochondriale semble se situer au carrefour d’un ensemble de processus interdépendants : contrôle de qualité de l’organite biogenèse, mitophagie, maintien de l’ADN mitochondrial -, fonctions bioénergétiques, distribution cellulaire et interaction avec les autres composants cellulaires, flux calciques, intégrations des voies de signalisation cellulaire et apoptose (→).
(→) Voir l’article de Jean-Michel Vallat et Benoît Funalot, page 842 de ce numéro
(→) Voir l’article de Cécile Sauvanet et al., page 823 de ce numéro
Les pathologies associées aux mutations d’OPAl, paradigmes des pathologies de la dynamique mitochondriale
Présentations cliniques
L’atrophie optique autosomique dominante (AOAD), initialement décrite en 1959 par l’ophtalmologiste danois Poul Kjer [7], est une neuropathie optique isolée, débutant de manière insidieuse au cours des deux premières décennies de la vie. L’atteinte simultanée des deux nerfs optiques aboutit à une perte du champ visuel central (scotome central), bilatérale et progressive, fréquemment associée à une difficulté de discrimination des couleurs dans les tons bleu-jaune (dyschromatopsie de type tritanopie). L’examen du fond d’œil révèle une pâleur temporale ou diffuse des papilles optiques (Figure 1). L’AOAD a une prévalence d’environ 1/50 000 individus, une pénétrance incomplète (43 à 90 % selon les études) et une expressivité clinique très hétérogène, allant d’une mal-vision à la cécité légale1.
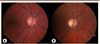 |
Figure 1 Atrophie optique : aspects au fond d’œil. A. Aspect normal de la tête du nerf optique droit visible au fond d’œil. B. Fond d’œil droit révélant une pâleur papillaire chez un patient atteint d’AOAD avec une acuité visuelle de 20/200. |
Les rares études histopathologiques post-mortem publiées décrivent une perte sélective des cellules ganglionnaires de la rétine et de leurs axones à l’origine des fibres du nerf optique. Plus récemment, la tomographie de cohérence optique in vivo2 a confirmé l’amincissement de la couche des fibres optiques autour de la papille optique chez les patients, alors que la rétine apparaît inaltérée [8].
Le dépistage moléculaire systématique des mutations d’OPA1 chez les patients atteints de différentes formes d’atrophie optique a révélé un très large éventail de variations phénotypiques associées aux mutations de ce gène. Une étude récente portant sur 104 patients qui appartiennent à 45 familles non apparentées a résumé les nouvelles formes cliniques décrites au cours de ces dix dernières années [9]. Près de 20 % des patients présentent une atteinte extra-oculaire associée à l’atrophie optique, ces formes cliniques étant qualifiées d’« AOAD plus » (Tableau I). L’atteinte la plus fréquente est une surdité neurosensorielle bilatérale et progressive débutant à l’adolescence. La seconde plus fréquente est un syndrome associant une ataxie, une myopathie, une neuropathie périphérique et une ophtalmoplégie chronique progressive externe3. Enfin, plus rarement, d’autres atteintes mimant la paraparésie spastique héréditaire ou la sclérose en plaques ont été rapportées. Il faut noter que ce large éventail de symptômes est superposable à celui qui est observé dans les pathologies classiquement associées à un déficit de la chaîne respiratoire mitochondriale.
Pathologies héréditaires dues à des mutations dans les gènes gouvernant les processus de fusion et de fission mitochondriales. OPAl : optic atrophy 1 ; MFN2 : mitofusine 2 ; GDAPl : ganglioside-induced differentiation-associated protein 1 ; CMT : Charcot-Marie-Tooth ; AOAD : atrophie optique autosomique dominante ; DRP1 : dynamin-related protein 1.
Le gène OPA1 et ses mutations
Le gène OPA1, long de plus de 100 kb, est composé de 31 exons dont 3 subissent un épissage alternatif (4, 4b et 5b) à l’origine de 8 isoformes protéiques [10]. Depuis l’identification du gène, 213 mutations pathogènes ont été rapportées, affectant l’ensemble des exons mais majoritairement ceux codant les domaines GTPase et dynamine4 [11]. Près de la moitié de ces mutations entraînent un arrêt prématuré de la traduction de la protéine. Aucune corrélation génotype-phénotype n’a pu être établie, sauf pour une mutation faux-sens du domaine GTPase (p.R445H) clairement associée aux formes avec neuropathies auditives et aux formes sévères multi-systémiques. La nature des mutations indique un mécanisme d’haplo-insuffisance pour les formes isolées d’AOAD et un effet dominant négatif pour les formes sévères souvent liées à des mutations faux-sens.
Hypothèses physiopathologiques
En accord avec l’implication d’OPA1 dans la fusion du réseau mitochondrial [12], nous avons montré que les fibroblastes de patients pouvaient présenter une altération de la dynamique membranaire avec un réseau fragmenté apparaissant sous forme de mitochondries ponctiformes (Figure 2) [13]. Il semblerait que cette fragmentation soit corrélée à la sévérité de la maladie puisqu’on la retrouve chez tous les patients syndromiques, alors que ce n’est pas systématiquement le cas chez les patients non syndromiques. En revanche, I’ensemble des lignées de fibroblastes étudiées présentent une susceptibilité accrue à l’apoptose [10] et une altération du fonctionnement de la chaîne respiratoire liée a un déficit d’activité soit du complexe I [14], soit du complexe IV [13]. De la même façon que pour l’atteinte de l’activité fusionnelle, il existe une bonne corrélation entre le niveau du déficit énergétique et la gravité de l’atteinte clinique. L’association de ces trois types d’altération contribue sans doute à la souffrance neuronale observée chez les patients. Les cellules ganglionnaires de la rétine sont tout particulièrement vulnérables du fait de l’absence de myélinisation de leur axone jusqu’à leur sortie du globe oculaire. Ces axones sont par conséquent directement exposés au stress induit par les rayons ultra-violets et requièrent un apport énergétique extrême pour transduire les potentiels d’action en l’absence de nœuds de Ranvier5. L’abondance mitochondriale, qui est frappante à la partie proximale de ces axones, permet probablement de répondre à ce besoin énergétique et explique en partie pourquoi l’atteinte des cellules ganglionnaires de la rétine est si fréquente dans les maladies mitochondriales.
 |
Figure 2 Morphologie du réseau mitochondrial dans un fibroblaste contrôle (A) (réseau tubulaire et interconnecté) et dans un fibroblaste porteur d’une mutation hétérozygote du gène OPA1 (B) (réseau fragmenté). |
La caractéristique la plus surprenante et inattendue observée chez les patients porteurs d’une mutation d’OPA1 est la présence de délétions multiples de l’ADN mitochondrial dans les muscles squelettiques [15]. Cette observation révèle pour la première fois le rôle de la dynamique mitochondriale dans le maintien de l’intégrité du génome mitochondrial alors que jusque-là, les pathologies dues à une instabilité de l’ADN mitochondrial étaient systématiquement en relation avec le système de réplication de ce génome [16].
Malgré la description de deux modèles murins mimant des mutations humaines et reproduisant les principales caractéristiques de la maladie [17, 18], il demeure difficile à ce jour de conclure de manière précise sur les mécanismes responsables de la maladie tant les rôles joués par OPA1 au sein de la mitochondrie sont multiples et pléiotropes.
Mitofusine 2 et maladie de Charcot-Marie-Tooth de type 2A
Présentations cliniques
En 2004, un second gène, MFN2 ou mitofusine 2, impliqué lui aussi dans la fusion mitochondriale, a été asso cié à une forme axonale à transmission autosomique dominante (CMT2A) de la maladie de Charcot-MarieTooth [3] (→). La maladie de Charcot-Marie-Tooth, qui regroupe des neuropathies périphériques héréditaires sensitivo-motrices fréquentes (1/2 500), a été découverte par trois médecins qui l’ont décrite comme étant « une forme particulière d’atrophie musculaire progressive, souvent familiale, débutant par les pieds et les jambes et atteignant plus tard les mains » [19, 20]. La maladie est caractérisée par un déficit moteur et une atrophie musculaire touchant les extrémités des membres, une réduction ou une abolition des réflexes ostéotendineux et des troubles sensitifs distaux, souvent associés à des déformations orthopédiques telles que des pieds creux. Son expressivité est très variable et son évolution ascendante est lente et progressive. Plusieurs formes sont décrites selon le mode de transmission, la valeur de la vitesse de conduction nerveuse et la classification moléculaire. Les formes axonales (CMT2) correspondent à des lésions prépondérantes de l’axone avec maintien de l’intégrité de la gaine de myéline. Une autre forme de CMT (hereditary motor and sensory neuropathy type VI, HMSN VI), qui associe neuropathies périphérique et optique, s’est révélée elle aussi due à des mutations de MFN2 [21]. Ainsi, la convergence entre la neuropathie périphérique retrouvée chez des patients AOAD plus et la présence d’une neuropathie optique chez certains patients MFN2 témoigne de mécanismes physiopathologiques communs à ces deux maladies de la fusion mitochondriale.
(→) Voir l’article de Jean-Michel Vallat et Benoît Funalot, page 842 de ce numéro
Localisé en 1p36.22, le gène MFN2 comporte 19 exons. Environ 60 mutations, majoritairement faux-sens, ont été rapportées. Elles affectent le plus souvent le domaine GTPase et les domaines d’interaction protéineprotéine qui permettent l’accolement entre les mitofusines de mitochondries adjacentes lors de I’initiation des évènements de fusion [22].
Hypothèses physiopathologiques
L’étude de biopsies nerveuses de patients ou de souris ayant une mutation de MFN2 montre une altération de la morphologie mitochondriale dans les axones. Les mitochondries apparaissent arrondies, de petite taille et souvent agrégées aux extrémités axonales et autour du noyau, suggérant une altération de leur fusion et de leur transport axonal [22, 23]. De plus, dans le nerf sciatique d’un modèle murin exprimant la mutation pathogène R94Q et la symptomatologie associées à la maladie, les axones de faible diamètre sont surreprésentés avec une augmentation de 30 % du nombre des mitochondries dans leur partie distale [24]. L’altération du trafic axonal est une hypothèse séduisante pour expliquer la vulnérabilité particulière des axones les plus longs de l’organisme, qui sont sélectivement affectés dans cette maladie. Cette hypothèse est confirmée par une étude récente montrant que les mitofusines jouent un rôle direct dans la régulation du transport axonal des mitochondries [25]. Une altération du métabolisme énergétique participe aussi très probablement à l’histoire de la maladie. En effet, MFN2 joue un rôle-clé dans la régulation du potentiel de membrane mitochondrial et de l’oxydation du glucose, du pyruvate et du palmitate. La baisse de l’expression de MFN2 est associée à une réduction de l’activité respiratoire mitochondriale dans les cellules musculaires squelettiques de patients atteints d’obésité ou de diabète de type 2. Inversement, la surexpression de MFN2 dans les cellules musculaires conduit à une augmentation du potentiel de membrane, de l’oxydation du glucose et de l’expression des sous-unités des complexes de la chaîne respiratoire ; elle s’observe dans le muscle squelettique des patients après un exercice physique et lors d’une perte de poids. Les fibroblastes de patients MFN2 atteints de CMT2A présentent une baisse d’efficacité des phosphorylations oxydatives et une diminution du potentiel de membrane mitochondrial, mais sans altération majeure de la morphologie mitochondriale, ce qui suggère que les rôles de MFN2 dans la fusion mitochondriale et dans la régulation du métabolisme énergétique sont indépendants [26]. Enfin, les mitofusines semblent jouer un rôle-clé dans le maintien de l’ADN mitochondrial [27]. Leur inactivation est en effet à l’origine, dans le muscle squelettique d’un modèle murin conditionnel dépourvu de mitofusines 1 et 2, d’une déplétion sévère du génome mitochondrial et d’une accumulation anormale de mutations. Les échanges de matériel génétique impliquant les processus de fusion interviennent donc très probablement dans la stabilité de l’ADN mitochondrial.
Mutation de DRP1 dans un syndrome neurologique sévère
En 2007, une mutation a aussi été détectée dans le gène DRP1 (dynamin-related protein 1), le troisième acteur majeur de la dynamique mitochondriale, impliqué dans la fission des mitochondries, chez un nouveau-né décédé à l’âge de 6 mois présentant un syndrome très sévère associant une microcéphalie, des anomalies du développement cérébral, une atrophie optique, une hypoplasie et une acidose lactique [4]. Les fibroblastes de ce nouveau-né présentaient un réseau mitochondrial et des peroxysomes hyperfusionnés, la particularité de DRP1 étant de participer à la fission de ces deux organites. À ce jour, aucun autre cas n’a été rapporté.
GDAP1 et maladies de Charcot-Marie-Tooth de type CMT2K et CMT4A
Enfin, le gène GDAP1, lui aussi acteur de la fission mitochondriale [28], a été impliqué, en 2002, dans une forme autosomique récessive démyélinisante de la maladie de Charcot-Marie-Tooth CMT4A [5] et en 2005, dans une forme autosomique dominante axonale CMT2K [6]. Ce gène, localisé en 8q13.1-12.3, possède six exons et code une protéine localisée à la membrane externe mitochondriale. Là encore, des anomalies de structure des mitochondries de type gonflement et une perturbation du métabolisme énergétique ont été mises en évidence chez les patients [29].
Un rôle ubiquitaire des perturbations de la dynamique mitochondriale dans les processus neurodégénératifs ?
La description progressive de ces différentes pathologies héréditaires confirme la très grande sensibilité des neurones du système nerveux central et périphérique aux dysfonctions mitochondriales et suggère que les anomalies de la plasticité mitochondriale pourraient être un élément important de la physiopathologie des affections neurodégénératives. Ce concept est renforcé par le fait que la sénescence dans les cellules post-mitotiques s’accompagne d’une modification profonde de la plasticité mitochondriale associant une élongation des mitochondries, une augmentation du nombre de connexions entre les tubules mitochondriaux et une dynamique mitochondriale altérée qui pourrait compromettre la maintenance de l’organite ainsi que sa résistance aux stress [30]. D’autre part, plusieurs études récentes ont démontré la présence d’anomalies de la morphologie mitochondriale dans les modèles des maladies de Parkinson, d’Alzheimer et de Huntington. Les protéines PINK1 (PTEN-induced kinase 1) et Parkine, responsables de formes héréditaires de la maladie de Parkinson, pourraient participer au contrôle de l’appareil de fusion/fission [31] par un mécanisme d’ubiquitinylation aboutissant à la destruction des mitochondries altérées [32]. L’atteinte de ce mécanisme de contrôle de qualité des mitochondries conduirait à une accumulation de mitochondries anormales et à la pathologie. Par ailleurs, le précurseur de la protéine amyloïde, par le biais de l’accumulation d’amyloïde-β, entraîne une fragmentation mitochondriale et un déséquilibre entre les processus de fission et de fusion en relation avec une diminution d’expression de la protéine OPA1 [33]. Enfin, la protéine Huntingtine mutée par expansion de polyglutamines induit aussi une fragmentation mitochondriale [34]. Ces trois maladies sont toutes associées à des anomalies du métabolisme énergétique mitochondrial connues de longue date [35]. Il apparaît donc que le paramètre énergétique mitochondrial est intimement lié à la structure et à la plasticité de l’organite. Cette étroite interrelation structure-fonction est donc sans doute à considérer comme jouant un rôle majeur dans les processus neurodégénératifs.
Conclusion et perspectives
En revenant au sens de notre démarche scientifique initiale qui nous a amenés à étudier l’évolution de la dynamique mitochondriale de la levure jusqu’aux pathologies humaines, quelles conclusions pouvons-nous retenir de ces dix années d’investigations ? Pour les organismes unicellulaires, la dynamique mitochondriale est un mécanisme déjà complexe qui nous a amenés à conceptualiser les mitochondries non pas comme un ensemble de particules indépendantes, mais comme un réseau unique qui requiert des échanges permanents pour homogénéiser les contenus matriciels, mais aussi pour éliminer les unités mitochondriales déficientes et ainsi orienter la mitochondriogenèse vers les unités mitochondriales énergétiquement les plus efficaces. Chez l’homme, ces mécanismes ont perduré et des contraintes supplémentaires s’y sont ajoutées pour répondre à des défis cellulaires extraordinaires, comme celui de transporter et distribuer les mitochondries le long des axones. La structure hautement spécialisée des neurones et leur consommation énergétique considérable les rend particulièrement vulnérables aux dysfonctionnements mitochondriaux. Ainsi, les neurones dont les axones sont les plus longs et ceux qui sont les plus gros consommateurs d’énergie se trouvent en première ligne dans les déficits héréditaires de la dynamique mitochondriale. Dans les pathologies neurodégénératives en relation avec le vieillissement, c’est le contrôle de qualité des mitochondries, lui-même sous la dépendance de la dynamique mitochondriale, qui pourrait être défaillant et conduire à l’accumulation progressive et délétère de mitochondries anormales. Bien que séduisantes, ces hypothèses doivent être fermement démontrées, puis résolues en termes de thérapeutique.
Conflit d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts concernant les données publiées dans cet article.
Remerciements
Les auteurs tiennent à remercier les membres des groupes d’Angers et de Montpellier pour leur contribution à ces travaux. Nous remercions également les associations de patients pour leur confiance et leur soutien : l’Union nationale des aveugles et déficients visuels (UNADEV), Rétina France, l’Association contre les maladies mitochondriales (AMMi), Ouvrir les yeux (OLY), et Forskningsrådet for Sundhed og Sygdom, Danemark.
Dans la plupart des pays anglo-saxons et plusieurs pays européens, on parle de cécité légale lorsque l’acuité visuelle du meilleur oeil est inférieure ou égale à 1/10 et de malvoyance lorsque l’acuité visuelle est inférieure à 5/10. En cas de cécité légale, la législation du pays prévoit une aide en faveur du patient.
La tomographie par cohérence optique est comparable dans son principe à l’imagerie ultrasonore; elle est basée sur la réflexion d’ondes non pas ultrasonores mais lumineuses.
Ophtalmoplégie chronique progressive externe : les ophtalmoplégies chroniques sont des affections caractérisées par une faiblesse progressive des muscles moteurs des globes oculaires et du releveur de la paupière supérieure (source orphanet).
Nœuds de Ranvier : interruptions régulières de la gaine de myéline entourant les axones, permettant ainsi la conduction saltatoire (donc plus rapide) de l’influx nerveux.
Références
- Delettre C, Lenaers G, Griffoin JM, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 2000 ; 26 : 207-10. [Google Scholar]
- Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynaminrelated GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 2000 ; 26 : 211-5. [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 2004 ; 36 : 449-51. [Google Scholar]
- Waterham HR, Koster J, Van Roermund CW, et al. A lethal defect of mitochondrial and peroxisomal fission. N Eng J Med 2007 ; 356 : 1736-41. [Google Scholar]
- Baxter RV, Ben Othmane K, Rochelle JM, et al. Ganglioside-induced differentiation-associated protein1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet 2002 ; 30 : 21-2. [Google Scholar]
- Claramunt R, Pedrola L, Sevilla T, et al. Genetics of Charcot-Marie-Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect. J Med Genet 2005 ; 42 : 358-65. [Google Scholar]
- Kjer P. Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol 1959 ; 164 (suppl 54) : 1-147. [Google Scholar]
- Milea D, Sander B, Wegener M, et al. Axonal loss occurs early in dominant optic atrophy. Acta Ophthalmol 2010 ; 88 : 342-6. [Google Scholar]
- Yu-Wai-Man P, Griffiths PG, Gorman GS, et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010 ; 133 : 771-86. [Google Scholar]
- Lenaers G, Reynier OPE1 Achouri G, et al. OPA1 functions in mitochondria and dysfunctions in optic nerve. Int J Biochem Cell Biol 2009 ; 41 : 1866-74. [Google Scholar]
- Ferré M, Amati-Bonneau P, Tourmen Y, et al. eOPA1: an online database for OPA1 mutations. Hum Mutat 2005 ; 25 : 423-8. [Google Scholar]
- Sauvanet C, Arnauné-Pelloquin L, David C, et al. Mitochondrial morphology and dynamics: actors, mecanisms and functions. Med Sci (Paris) 2010 ; 26 : 823-9. [Google Scholar]
- Chevrollier A, Guillet V, Loiseau D, et al. Hereditary optic neuropathies share a common mitochondrial coupling defect. Ann Neurol 2008 ; 63 : 794-8. [Google Scholar]
- Zanna C, Ghelli A, Porcelli AM, et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 2008 ; 131 : 352-67. [Google Scholar]
- Amati-Bonneau P, Valentino ML, Reynier P, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy plus phenotypes. Brain 2008 ; 131 : 338-51. [Google Scholar]
- Sarzi E, Rötig A. Instabilité du génome mitochondrial et pathologies associées. Med Sci (Paris) 2010 ; 26 : 171-6. [Google Scholar]
- Alavi MV, Bette S, Schimpf S, et al. A splice site mutation in the murine OPA1 gene features pathology of autosomal dominant optic atrophy. Brain 2007 ; 130 : 1029-42. [Google Scholar]
- Davies VJ, Hollins AJ, Piechota MJ, et al. OPA1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet 2007 ; 16 : 1307-18. [Google Scholar]
- Charcot JM, Marie P. Sur une forme particulière d’atrophie musculaire progressive souvent familiale débutant par les pieds et les jambes et atteignant plus tard les mains. Rev Med 1886 ; 6 : 97-138. [Google Scholar]
- Tooth H. The peroneal type of progressive muscular atrophy. London : HK Lewis, 1886. [Google Scholar]
- Züchner S, De Jonghe P, Jordanova A, et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol 2006 ; 59 : 276-81. [Google Scholar]
- Cartoni R, Martinou JC. Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol 2009 ; 218 : 268-73. [Google Scholar]
- Vallat JM, Ouvrier RA, Pollard JD, et al. Histopathological findings in hereditary motor and sensory neuropathy of axonal type with onset in early childhood associated with mitofusin 2 mutations. J Neuropathol Exp Neurol 2008 ; 67 : 1097-102. [Google Scholar]
- Cartoni R, Arnaud E, Médard JJ, et al. Expression of mitofusin 2(R94Q) in a transgenic mouse leads to Charcot-Marie-Tooth neuropathy type 2A. Brain 2010 ; 133 : 1460-9. [Google Scholar]
- Misko A, Jiang S, Wegorzewska I, et al. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci 2010 ; 30 : 4232-40. [Google Scholar]
- Loiseau D, Chevrollier A, Verny C, et al. Mitochondrial Coupling Defect in Charcot-Marie-Tooth Type 2A disease. Ann Neurol 2007 ; 61 : 315-23. [Google Scholar]
- Chen H, Vermulst M, Wang YE, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010 ; 141 : 280-9. [Google Scholar]
- Niemann A, Ruegg M, La Padula V, et al. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol 2005 ; 170 : 1067-78. [Google Scholar]
- Cassereau J, Chevrollier A, Gueguen N, et al. Mitochondrial complex I deficiency in GDAP1-related autosomal dominant Charcot-Marie-Tooth disease (CMT2K). Neurogenetics 2009 ; 10 : 145-50. [Google Scholar]
- Mai S, Klinkenberg M, Auburger G, et al. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J Cell Sci 2010 ; 123 : 917-26. [Google Scholar]
- Yang Y, Ouyang Y, Yang L, et al. Pink1 regulates mitochondrial dynamics throught interaction with the fission/fusion machinery. Proc Natl Acad Sci USA 2008 ; 105 : 7070-5. [Google Scholar]
- Ziviani E, Tao RN, Whitworth AJ. Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquinates mitofusin. Proc Natl Acad Sci USA 2010 ; 107 : 5018-23. [Google Scholar]
- Wang X, Su B, Siedlak SL, et al. Amyloid beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA 2008 ; 105 : 19318-23. [Google Scholar]
- Wang H, Lim PJ, Karbowski M, Monteiro MJ. Effects of overexpression of Huntingtin proteins on mitochondrial integrity. Hum Mol Genet 2008 ; 18 : 737-52. [Google Scholar]
- Fukui H, Moraes CT. The mitochondrial impairment, oxidative stress and neurodegeneration connection: reality or just an attractive hypothesis? Trends Neurosci 2008 ; 31 : 251-6. [Google Scholar]
- Vallat JM, Funalot B. La maladie de Charcot-Marie-Tooth. Med Sci (Paris) 2010 ; 26 : 842-7. [Google Scholar]
Liste des tableaux
Pathologies héréditaires dues à des mutations dans les gènes gouvernant les processus de fusion et de fission mitochondriales. OPAl : optic atrophy 1 ; MFN2 : mitofusine 2 ; GDAPl : ganglioside-induced differentiation-associated protein 1 ; CMT : Charcot-Marie-Tooth ; AOAD : atrophie optique autosomique dominante ; DRP1 : dynamin-related protein 1.
Liste des figures
|
Figure 1 Atrophie optique : aspects au fond d’œil. A. Aspect normal de la tête du nerf optique droit visible au fond d’œil. B. Fond d’œil droit révélant une pâleur papillaire chez un patient atteint d’AOAD avec une acuité visuelle de 20/200. |
| Dans le texte | |
|
Figure 2 Morphologie du réseau mitochondrial dans un fibroblaste contrôle (A) (réseau tubulaire et interconnecté) et dans un fibroblaste porteur d’une mutation hétérozygote du gène OPA1 (B) (réseau fragmenté). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.